Next-MP50 Stats Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
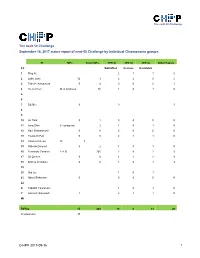
Next-MP50 Status Report
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

Assembly and Analysis of 100 Full MHC Haplotypes from the Danish
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press 1 Assembly and analysis of 100 full MHC haplotypes from the Danish 2 population 3 4 Jacob M. Jensen1*, Palle Villesen1,2, Rune M. Friborg1, The Danish Pan 5 Genome Consortium, Thomas Mailund1, Søren Besenbacher1,3, Mikkel H. 6 Schierup1,4* 7 8 1. Bioinformatics Research Centre, Aarhus University, CF Møllers Alle 8, 9 8000 Aarhus C. Denmark 10 2. Department of Clinical Medicine, Aarhus University, Palle Juul-Jensens 11 Boulevard 82, 8200 Aarhus N. Denmark 12 3. Department of Molecular Medicine, Aarhus University Hospital, Skejby, 13 Aarhus N 8200, Denmark 14 4. Department of Bioscience, Aarhus University, Ny Munkegade 116, 8000 15 Aarhus C. Denmark 16 17 * Correspondence to [email protected], [email protected] 18 19 1 Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press 20 Abstract 21 Genes in the major histocompatibility complex (MHC, also known as 22 HLA) play a critical role in the immune response and variation within the 23 extended 4 Mb region shows association with major risks of many 24 diseases. Yet, deciphering the underlying causes of these associations 25 is difficult because the MHC is the most polymorphic region of the 26 genome with a complex linkage disequilibrium structure. Here we 27 reconstruct full MHC haplotypes from de novo assembled trios without 28 relying on a reference genome and perform evolutionary analyses. We 29 report 100 full MHC haplotypes and call a large set of structural variants 30 in the regions for future use in imputation with GWAS data. -

Large-Scale Evolutionary Analysis of Polymorphic Inversions in the Human Genome
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Doctoral thesis Large-scale evolutionary analysis of polymorphic inversions in the human genome Author Carla Giner Delgado Director Mario Caceres´ Aguilar Departament de Gen`eticai de Microbiologia Facultat de Bioci`encies Universitat Aut`onomade Barcelona 2017 Large-scale evolutionary analysis of polymorphic inversions in the human genome Mem`oriapresentada per Carla Giner Delgado per a optar al grau de Doctora en Gen`etica per la Universitat Aut`onomade Barcelona Autora Director Carla Giner Delgado Mario Caceres´ Aguilar Bellaterra, 28 de Setembre de 2017 Abstract Chromosomal inversions are structural variants that invert a fragment of the genome without usually modifying its content, and their subtle but powerful effects in natural populations have fascinated evolutionary biologists for a long time. Discovered a century ago in fruit flies, their association with dif- ferent evolutionary processes, such as local adaptation and speciation, was soon evident in several species. However, in the current era of genomics and big data, inversions frequently escape the grasp of current technologies and remain largely overlooked in humans. -

Evolution and Diversity of Copy Number Variation in the Great Ape Lineage
Downloaded from genome.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Evolution and diversity of copy number variation in the great ape lineage Peter H. Sudmant1, John Huddleston1,7, Claudia R. Catacchio2, Maika Malig1, LaDeana W. Hillier3, Carl Baker1, Kiana Mohajeri1, Ivanela Kondova4, Ronald E. Bontrop4, Stephan Persengiev4, Francesca Antonacci2, Mario Ventura2, Javier Prado-Martinez5, Tomas 5,6 1,7 Marques-Bonet , and Evan E. Eichler 1. Department of Genome Sciences, University of Washington, Seattle, WA, USA 2. University of Bari, Bari, Italy 3. The Genome Institute, Washington University School of Medicine, St. Louis, MO, USA 4. Department of Comparative Genetics, Biomedical Primate Research Centre, Rijswijk, The Netherlands 5. Institut de Biologia Evolutiva, (UPF-CSIC) Barcelona, Spain 6. Institució Catalana de Recerca i Estudis Avançats (ICREA), Barcelona, Spain 7. Howard Hughes Medical Institute, University of Washington, Seattle, WA, USA Correspondence to: Evan Eichler Department of Genome Sciences University of Washington School of Medicine Foege S-413A, Box 355065 3720 15th Ave NE Seattle, WA 98195 E-mail: [email protected] 1 Downloaded from genome.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Copy number variation (CNV) contributes to the genetic basis of disease and has significantly restructured the genomes of humans and great apes. The diversity and rate of this process, however, has not been extensively explored among the great ape lineages. We analyzed 97 deeply sequenced great ape and human genomes and estimate that 16% (469 Mbp) of the hominid genome has been affected by recent copy number changes. -

Transcriptome Analyses of Lncrnas in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links Between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa
antioxidants Article Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa Luigi Donato 1,2,* , Concetta Scimone 1,2 , Simona Alibrandi 1,3, Carmela Rinaldi 1, Antonina Sidoti 1,2,* and Rosalia D’Angelo 1,2 1 Department of Biomedical and Dental Sciences and Morphofunctional Imaging, Division of Medical Biotechnologies and Preventive Medicine, University of Messina, 98125 Messina, Italy; [email protected] (C.S.); [email protected] (S.A.); [email protected] (C.R.); [email protected] (R.D.) 2 Department of Biomolecular Strategies, Genetics and Avant-Garde Therapies, I.E.ME.S.T., 90139 Palermo, Italy 3 Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, 98125 Messina, Italy * Correspondence: [email protected] (L.D.); [email protected] (A.S.); Tel.: +39-0902213136 (L.D.); +39-0902213372 (A.S.) Received: 21 March 2020; Accepted: 14 April 2020; Published: 15 April 2020 Abstract: Long non-coding RNAs (lncRNAs) are untranslated transcripts which regulate many biological processes. Changes in lncRNA expression pattern are well-known related to various human disorders, such as ocular diseases. Among them, retinitis pigmentosa, one of the most heterogeneous inherited disorder, is strictly related to oxidative stress. However, little is known about regulative aspects able to link oxidative stress to etiopathogenesis of retinitis. Thus, we realized a total RNA-Seq experiment, analyzing human retinal pigment epithelium cells treated by the oxidant agent N-retinylidene-N-retinylethanolamine (A2E), considering three independent experimental groups (untreated control cells, cells treated for 3 h and cells treated for 6 h). -

Genetic Overlap Between Alzheimer&Rsquo
Molecular Psychiatry (2015) 20, 1588–1595 © 2015 Macmillan Publishers Limited All rights reserved 1359-4184/15 www.nature.com/mp ORIGINAL ARTICLE Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus RS Desikan1, AJ Schork2, Y Wang3,4, A Witoelar4, M Sharma5,6, LK McEvoy1, D Holland3, JB Brewer1,3, C-H Chen1,7, WK Thompson7, D Harold8, J Williams8, MJ Owen8,MCO’Donovan8, MA Pericak-Vance9, R Mayeux10, JL Haines11, LA Farrer12, GD Schellenberg13, P Heutink14, AB Singleton15, A Brice16, NW Wood17, J Hardy18, M Martinez19, SH Choi20, A DeStefano20,21, MA Ikram22, JC Bis23, A Smith24, AL Fitzpatrick25, L Launer26, C van Duijn22, S Seshadri21,27, ID Ulstein28, D Aarsland29,30,31, T Fladby32,33, S Djurovic4, BT Hyman34, J Snaedal35, H Stefansson36, K Stefansson36,37, T Gasser5, OA Andreassen4,7, AM Dale1,2,3,7for the ADNI38ADGC, GERAD, CHARGE and IPDGC Investigators39 We investigated the genetic overlap between Alzheimer’s disease (AD) and Parkinson’s disease (PD). Using summary statistics (P-values) from large recent genome-wide association studies (GWAS) (total n = 89 904 individuals), we sought to identify single nucleotide polymorphisms (SNPs) associating with both AD and PD. We found and replicated association of both AD and PD with the A allele of rs393152 within the extended MAPT region on chromosome 17 (meta analysis P-value across five independent AD cohorts = 1.65 × 10− 7). In independent datasets, we found a dose-dependent effect of the A allele of rs393152 on intra-cerebral MAPT transcript levels and volume loss within the entorhinal cortex and hippocampus. -

Kauer Washington 0250O 21245.Pdf (1.990Mb)
Using biomedical data to identify genetic variants that drive drug responses in Acute Myeloid Leukemia Nicole Kauer A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science University of Washington Tacoma 2020 Committee: Ka Yee Yeung-Rhee, Chair Ling-Hong Hung Wes Lloyd Program Authorized to Offer Degree: Computer Science and Systems c Copyright 2020 Nicole Kauer University of Washington Tacoma Abstract Using biomedical data to identify genetic variants that drive drug responses in Acute Myeloid Leukemia Nicole Kauer Chair of the Supervisory Committee: Professor Ka Yee Yeung-Rhee School of Engineering and Technology Acute Myeloid Leukemia (AML) is a heterogeneous cancer of the blood that progresses quickly, with approximately 10,000 AML related deaths reported annually in the United States. Patients with AML tend to have genetic variations, which can significantly affect drug sensitivity and treatment outcomes. While massive amounts of big biomedical data have been generated to characterize AML, these genetic variations are not yet well-understood. Thus, the development of individualized approaches to AML therapy using these big data has great potential. The promise of precision medicine is that knowledge of the genetic characteristics present within a cancer will enable better choices for therapy. In this thesis, we applied data sci- ence techniques to analyze AML biomedical data in collaboration with Dr. Pamela Becker, Hematology, UW-Seattle, with the goal of identifying genetic variants that drive drug re- sponses in AML. We identified 30 novel gene-drug pairs with statistical significant responses. Additionally, both univariate and multivariate machine learning models were created, with multivariate feature selection via Bayesian Model Averaging. -

Next-MP50 Status Report
The neXt-50 Challenge Status report and acceptance of next-50 Challenge by individual Chromosome groups February 24, 2017. Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). Status of the Chromosome “parts list”: Step by step milestone plan to find, identify and validate MPs 1. We try to identify more missing proteins through high coverage proteomics technology with testis samples. We will finish this project before May. 2. We tested and confirmed that PTM approach could identify significant number of missing proteins. We will finalize the data sets before May. C-HPP 2017-03-31 The neXt-50 Challenge Chromosome 2 See combined response and work plan with Chromosome 14. Chromosome 3 (Takeshi Kawamura) PIC Leaders: Takeshi Kawamura (since January 1) Contributing labs: Major lab members or partners contributing to the neXt50: Takeshi Kawamura: Associate Professor, Proteomics Laboratory, Isotope Science Center, The University of Tokyo, Tokyo, Japan. Toshihide Nishimura: Professor, Department of Translational Medicine Informatics, St. Marianna University School of Medicine, Kanagawa, Japan. Hiromasa Tojo: Associate Professor, Department of Biophysics and Biochemistry, Osaka University; Graduate School of Medicine, Osaka, Japan. -

Characterizing Genomic Duplication in Autism Spectrum Disorder by Edward James Higginbotham a Thesis Submitted in Conformity
Characterizing Genomic Duplication in Autism Spectrum Disorder by Edward James Higginbotham A thesis submitted in conformity with the requirements for the degree of Master of Science Graduate Department of Molecular Genetics University of Toronto © Copyright by Edward James Higginbotham 2020 i Abstract Characterizing Genomic Duplication in Autism Spectrum Disorder Edward James Higginbotham Master of Science Graduate Department of Molecular Genetics University of Toronto 2020 Duplication, the gain of additional copies of genomic material relative to its ancestral diploid state is yet to achieve full appreciation for its role in human traits and disease. Challenges include accurately genotyping, annotating, and characterizing the properties of duplications, and resolving duplication mechanisms. Whole genome sequencing, in principle, should enable accurate detection of duplications in a single experiment. This thesis makes use of the technology to catalogue disease relevant duplications in the genomes of 2,739 individuals with Autism Spectrum Disorder (ASD) who enrolled in the Autism Speaks MSSNG Project. Fine-mapping the breakpoint junctions of 259 ASD-relevant duplications identified 34 (13.1%) variants with complex genomic structures as well as tandem (193/259, 74.5%) and NAHR- mediated (6/259, 2.3%) duplications. As whole genome sequencing-based studies expand in scale and reach, a continued focus on generating high-quality, standardized duplication data will be prerequisite to addressing their associated biological mechanisms. ii Acknowledgements I thank Dr. Stephen Scherer for his leadership par excellence, his generosity, and for giving me a chance. I am grateful for his investment and the opportunities afforded me, from which I have learned and benefited. I would next thank Drs. -

Comparative Analysis of the Ubiquitin-Proteasome System in Homo Sapiens and Saccharomyces Cerevisiae
Comparative Analysis of the Ubiquitin-proteasome system in Homo sapiens and Saccharomyces cerevisiae Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Universität zu Köln vorgelegt von Hartmut Scheel aus Rheinbach Köln, 2005 Berichterstatter: Prof. Dr. R. Jürgen Dohmen Prof. Dr. Thomas Langer Dr. Kay Hofmann Tag der mündlichen Prüfung: 18.07.2005 Zusammenfassung I Zusammenfassung Das Ubiquitin-Proteasom System (UPS) stellt den wichtigsten Abbauweg für intrazelluläre Proteine in eukaryotischen Zellen dar. Das abzubauende Protein wird zunächst über eine Enzym-Kaskade mit einer kovalent gebundenen Ubiquitinkette markiert. Anschließend wird das konjugierte Substrat vom Proteasom erkannt und proteolytisch gespalten. Ubiquitin besitzt eine Reihe von Homologen, die ebenfalls posttranslational an Proteine gekoppelt werden können, wie z.B. SUMO und NEDD8. Die hierbei verwendeten Aktivierungs- und Konjugations-Kaskaden sind vollständig analog zu der des Ubiquitin- Systems. Es ist charakteristisch für das UPS, daß sich die Vielzahl der daran beteiligten Proteine aus nur wenigen Proteinfamilien rekrutiert, die durch gemeinsame, funktionale Homologiedomänen gekennzeichnet sind. Einige dieser funktionalen Domänen sind auch in den Modifikations-Systemen der Ubiquitin-Homologen zu finden, jedoch verfügen diese Systeme zusätzlich über spezifische Domänentypen. Homologiedomänen lassen sich als mathematische Modelle in Form von Domänen- deskriptoren (Profile) beschreiben. Diese Deskriptoren können wiederum dazu verwendet werden, mit Hilfe geeigneter Verfahren eine gegebene Proteinsequenz auf das Vorliegen von entsprechenden Homologiedomänen zu untersuchen. Da die im UPS involvierten Homologie- domänen fast ausschließlich auf dieses System und seine Analoga beschränkt sind, können domänen-spezifische Profile zur Katalogisierung der UPS-relevanten Proteine einer Spezies verwendet werden. Auf dieser Basis können dann die entsprechenden UPS-Repertoires verschiedener Spezies miteinander verglichen werden. -

Genomic Approach in Idiopathic Intellectual Disability Maria De Fátima E Costa Torres
ESTUDOS DE 8 01 PDPGM 2 CICLO Genomic approach in idiopathic intellectual disability Maria de Fátima e Costa Torres D Autor. Maria de Fátima e Costa Torres D.ICBAS 2018 Genomic approach in idiopathic intellectual disability Genomic approach in idiopathic intellectual disability Maria de Fátima e Costa Torres SEDE ADMINISTRATIVA INSTITUTO DE CIÊNCIAS BIOMÉDICAS ABEL SALAZAR FACULDADE DE MEDICINA MARIA DE FÁTIMA E COSTA TORRES GENOMIC APPROACH IN IDIOPATHIC INTELLECTUAL DISABILITY Tese de Candidatura ao grau de Doutor em Patologia e Genética Molecular, submetida ao Instituto de Ciências Biomédicas Abel Salazar da Universidade do Porto Orientadora – Doutora Patrícia Espinheira de Sá Maciel Categoria – Professora Associada Afiliação – Escola de Medicina e Ciências da Saúde da Universidade do Minho Coorientadora – Doutora Maria da Purificação Valenzuela Sampaio Tavares Categoria – Professora Catedrática Afiliação – Faculdade de Medicina Dentária da Universidade do Porto Coorientadora – Doutora Filipa Abreu Gomes de Carvalho Categoria – Professora Auxiliar com Agregação Afiliação – Faculdade de Medicina da Universidade do Porto DECLARAÇÃO Dissertação/Tese Identificação do autor Nome completo _Maria de Fátima e Costa Torres_ N.º de identificação civil _07718822 N.º de estudante __ 198600524___ Email institucional [email protected] OU: [email protected] _ Email alternativo [email protected] _ Tlf/Tlm _918197020_ Ciclo de estudos (Mestrado/Doutoramento) _Patologia e Genética Molecular__ Faculdade/Instituto _Instituto de Ciências -

C 2018 Sheng Wang LEVERAGING KNOWLEDGE NETWORKS for PRECISION MEDICINE
c 2018 Sheng Wang LEVERAGING KNOWLEDGE NETWORKS FOR PRECISION MEDICINE BY SHENG WANG DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Computer Science in the Graduate College of the University of Illinois at Urbana-Champaign, 2018 Urbana, Illinois Doctoral Committee: Professor ChengXiang Zhai, Chair Assistant Professor Jian Peng, Director of Research Professor Saurabh Sinha Professor Jiawei Han Professor Xinghua Lu ABSTRACT Akin to the exponential growth of genomic sequencing data, high-throughput techniques in proteomics and biotechnology have been creating ever-expanding repositories of proteomic, pharmacological, and interactomic data. Other molecular data, including expression profiles, genomic mutations and cell conditions, have also been massively generated and they are further refining our understanding of disease mechanisms. In addition, patient data, gathered by electronic medical record systems and social medias, complement biological data and pave the way for personalized treatment strategies. Therefore, efficiently and effectively integrating and mining these invaluable data hold the great promising of making precision medicine a reality. However, integrating and mining these large-scale, heterogeneous, and noisy dataset pose several fundamental computational challenges and have therefore become a bottleneck to clinical decision making and medical knowledge discovery. This thesis is a systematic study of mining these biological and healthcare data for precision medicine. I take a network perspective and integrate these datasets into a large knowledge network where nodes are biological concepts and links are biological relationships. I then propose a novel computa- tional framework to mine these knowledge networks. To demonstrate the effectiveness of mining knowledge networks, I will introduce how this framework can be used to understand molecular functions, accelerate drug discovery, and support clinical decision making.