Phosphate Binding by Sucroferric Oxyhydroxide Ameliorates Renal
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
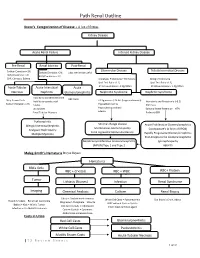
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium -

Management of the Nephrotic Syndrome GAVIN C
Arch Dis Child: first published as 10.1136/adc.43.228.257 on 1 April 1968. Downloaded from Personal Practice* Arch. Dis. Childh., 1968, 43, 257. Management of the Nephrotic Syndrome GAVIN C. ARNEIL From the Department of Child Health, University of Glasgow The nephrotic syndrome may be defined as gross (3) Idiopathic nephrosis. This may be re- proteinuria, predominantly albuminuria, with con- garded either as a primary disease, or as a secondary sequent selective hypoproteinaemia; usually accom- disorder, with the primary cause or causes still panied by oedema, ascites, and hyperlipaemia, and unknown. It is the common form of nephrosis in sometimes by systemic hypertension, azotaemia, or childhood, but the less common in adult patients. excessive erythrocyturia. Unless otherwise specified, the present discussion concerns idiopathic nephrosis. Grouping of Nephrotic Syndrome The syndrome falls into three groups. Assessment of the Case Full history and examination helps to exclude (1) Congenital nephrosis. A group of dis- many secondary forms of the disease. Blood orders present at and presenting soon after birth, pressure is recorded employing as broad a cuff as which are familial and may be acquired or inherited. practicable, and the width of the cuff used is Persistent oedema attracts attention in the first recorded as of importance in comparative estima- weeks of life. The disease may be rapidly lethal, tions. A careful search for concomitant infection but occasionally the patient will survive for a year is made, particularly to exclude pneumococcal copyright. or more without fatal infection or lethal azotaemia. peritonitis or low grade cellulitis. In a patient The disease pattern tends to run true within a previously treated elsewhere, evidence of steroid family. -

The Nephrotic Syndrome in Early Infancy: a Report of Three Cases* by H
Arch Dis Child: first published as 10.1136/adc.32.163.167 on 1 June 1957. Downloaded from THE NEPHROTIC SYNDROME IN EARLY INFANCY: A REPORT OF THREE CASES* BY H. McC. GILES, R. C. B. PUGH, E. M. DARMADY, FAY STRANACK and L. I. WOOLF From the Department of Paediatrics, St. Mary's Hospital, The Hospital for Sick Children, Great Ormond Street, London, and the Central Laboratory, Portsmouth (RECEIVED FOR PUBLICATION, DECEMBER 12, 1956) The nephrotic syndrome is characterized by evidence of genetic aetiology inasmuch as two of generalized oedema, hypoproteinaemia with diminu- the infants were brother and sister and both sets of tion or reversal of the albumin/globulin ratio, hyper- parents were cousins. Further points of interest lipaemia and gross albuminuria. Haematuria is not were the demonstration of anisotropic crystalline prominent; hypertension and azotaemia, except in material in alcohol-fixed tissues from all three the terminal stages, are slight and transient and infants, and the discovery on renal microdissection often do not occur at all. Recognition of this of lesions of the proximal tubule recalling those syndrome presents no difficulty but in many cases, found in Fanconi-Lignac disease (cystinosis). particularly in childhood, its cause remains obscure. Occasionally certain drugs such as troxidone or mercury, or certain diseases such as amyloidosis, Case Reports pyelonephritis, diabetes, disseminated lupus erythe- The parents of the first two patients (and the paternal copyright. matosus or renal vein thrombosis, can be in- grandparents) are first cousins. They, and a sister born criminated. In cases which develop renal failure three years before the first of her affected siblings, have and come to necropsy the lesions of glomerulo- been investigated in detail and show no sign of renal II disease. -

Kidney Disease: the Basics
Kidney Disease: The Basics Note: Footnotes in body text may not run sequentially due to ongoing updates. However, they do correspond correctly to the numbers in the reference list. Fast Facts Kidney disease, also known as chronic kidney disease or CKD, causes more deaths than breast cancer or prostate cancer (NVS 2021 report of 2018 data).1 It is the under-recognized public health crisis. • Kidney disease affects an estimated 37 million people in the U.S. (15% of the adult population; more than 1 in 7 adults).2,3,4 • Approximately 90% of those with kidney disease don’t know they have it.2 • And 2 of 5 adults with severe kidney disease don’t know they have it.2 • 1 in 3 adults in the U.S. (approximately 80 million) is at risk for kidney disease.2,5 • Kidney disease is more common in women (14%) than men (12%).2 But for every 2 women who develop end- stage kidney disease (ESKD), 3 men’s kidneys fail.2 • Kidney disease is a leading cause of death in the U.S.1,6,7 • About 1 in 2 people with very low kidney function (not on dialysis) don’t know they have kidney disease.2,13 • Approximately 1 in 3 adults with diabetes and 1 in 5 adults with high blood pressure may have kidney disease.2 • COVID-19 is targeting people with kidney disease, kidney transplant patients, and those at risk for kidney disease. [See www.kidney.org/covid-19] What is Kidney Disease? Chronic kidney disease (CKD) means your kidneys are damaged and losing their ability to keep you healthy by filtering your blood. -

Chronic Kidney Disease in South Carolina
Chronic Kidney Disease in South Carolina WHAT IS KIDNEY DISEASE1? KIDNEY DISEASE IN S.C. • Chronic Kidney Disease (CKD) reduces the body’s ability Mortality to filter blood, remove waste and extra water, and keep beneficial electrolytes in balance. • 868 people died from Kidney Disease in 2013 (most recent data available). The mortality rate decreased from • Left untreated, CKD can lead to kidney failure. 19.8 in 2010 to 16.0 in 2013 but still remains above the • When this happens, dialysis or a kidney transplant is the national rate. only option for keeping a person alive. • Deaths from Nephritis, Nephrotic Syndrome, and • Early kidney disease is a silent problem! Nephrosis are the 8th leading cause of death. U.S. STATISTICS • More than 20 million Americans have chronic kidney disease1. • Many more Americans, including anyone with high blood pressure, diabetes, or a family history of kidney disease, are at an increased risk2. • The number of people developing kidney failure has doubled each decade for the last two decades3. • End-Stage Renal Disease (ESRD) resulted in 88,638 deaths in 20124. • Treating Medicare patients aged 65 or older with kidney • Mortality from Kidney Disease for blacks in South disease cost the United States $44.6 billion in 20125. Carolina is more than twice as high as whites. • 47,112 people died from Nephritis, Nephrotic Syndrome, and Nephrosis in 2013 (14.9 per 100,000 population). This was the 9th leading cause of death6. WHO IS AT RISK1? • Diabetes is the most common cause of kidney failure. Approximately 1 out of 3 adults with diabetes has CKD. -

A Clinical and Pathological Study of Schistosomal Nephritis*
Bull. Org. mond. Sante 1972, 47, 549-557 Bull. Wld Hlth Org. A clinical and pathological study of schistosomal nephritis* M. S. SABBOUR,1 W. EL-SAID,' & I. ABOU-GABAL3 In a Cairo clinic 17 of41patients with chronicpyelonephritis secondary to urinary schisto- somiasis presented with classicalfeatures of the nephrotic syndrome, two-thirds being hyper- tensive and the majority having glomerular filtration rates within the normal range. Hyper- cholesterolaemia was found in one-third of the patients. Urinary sediments from these patients contained a preponderance of pus cells, red cells, granular casts, or pus casts. In addition to patches ofpyelonephritis, the glomeruli showed diffuse andfocal glomerulo- sclerosis. Electron microscopy revealed basement-membrane-like deposits in the hypertro- phied axial endothelial cells and electron-dense deposits along the glomerular basement membrane. This variety of nephrotic syndrome associated with schistosomal pyelonephritis was the most common cause of nephrotic syndrome seen in the clinic. During the last few years, we have had the impres- urine analysis. A modified Addis technique (El-Said, sion that patients with urinary schistosomiasis and 1971) was used to count the exact number of pus secondary bacterial pyelonephritis develop a neph- cells excreted per hour in urine. rotic syndrome. The majority of patients admitted Only confirmed cases of chronic pyelonephritis to the renal unit are suffering from chronic pyelo- were included in this study, doubtful cases being nephritis secondary to urinary schistosomiasis (Abou- excluded. Patients with, in addition to the pyelo- Gabal et al., 1970), and a considerable proportion nephritis, other lesions that could give rise to the of them have varying degrees of generalized oedema. -

The Impact of Chronic Kidney Disease in West Virginia
THE IMPACT OF CHRONIC KIDNEY DISEASE IN WEST VIRGINIA Joe Manchin III Governor Martha Yeager Walker Secretary Department of Health and Human Resources April 2006 West Virginia Bureau for Public Health Chris Curtis, MPH Acting Commissioner Catherine Slemp, MD, MPH Acting Medical Officer Joe Barker, MPA Director, Office of Epidemiology and Health Promotion Daniel M. Christy, MPA Director, Health Statistics Center Report Written By Eugenia Thoenen West Virginia Kidney Advisory Group Peggy J. Adams, MSN, RN, CDE Daniel M. Christy, MPA James C. Doria Mary Emmett, PhD Marie Gravely, MA, RD, LD, CDE Derrick Latos, MD, MACP Tammie Mitchell, RN, BSN, CNN Cecil Pollard, MA Rebecca Schmidt, DO, FACP, FASN Henry Taylor, MD, MPH Jessica Wright, RN, MPH Health Statistics Center James C. Doria, Program Manager, Statistical Services Unit Fred King, BRFSS Coordinator Thomas A. Leonard, MS, Programmer/Analyst Tom Light, Programmer Additional Acknowledgments Jay Eckhart, Health Data Analyst West Virginia Health Care Authority ii Executive Summary General Facts ● The kidneys are the chemists of our bodies. They have three main functions: (1) remove waste products, (2) balance our bodies’ chemicals, and (3) produce essential hormones. ● The kidneys filter approximately 50 gallons of blood every day; in general, a healthy adult will eliminate approximately one to two quarts of urine daily. If the kidneys do not function properly, wastes accumulate in the body. The progressive loss of kidney function eventually leads to end-stage renal disease (ESRD), or kidney failure, resulting in the need for either dialysis or kidney transplantation. ● The National Kidney Foundation (NKF) estimates that 1 in every 9 adults in the United States, more than 20 million people, has chronic kidney disease (CKD) and that more than 20 million others are at increased risk for the disease. -

A Case of Mononucleosis Infectiosa Presenting with Cholemic Nephrosis
NDT Plus (2011) 4: 170–172 doi: 10.1093/ndtplus/sfr038 Advance Access publication 14 April 2011 Case Report A case of mononucleosis infectiosa presenting with cholemic nephrosis Obbo W. Bredewold, Johan W. de Fijter and Ton Rabelink Department of Nephrology, Leiden University Medical Center, Albinusdreef 2, 2333 ZA Leiden, The Netherlands Correspondence and offprint requests to: Obbo Bredewold; E-mail: [email protected] Abstract examination revealed jaundice, besides enlarged and painful A 38-year-old male was admitted with fever, progressive cervical lymph nodes. Fine crackles were heard over both jaundice, cervical lymphadenopathy, hepatomegaly and lungs, and the spleen was slightly enlarged. The remainder acute oliguric renal failure. Epstein-Barr virus (EBV) infec- of the investigation was normal. After laboratory examina- tion was diagnosed by detection of EBV-DNA in plasma tion (Table 1), acute EBV infection with obstructive jaundice and confirmed by EBV seroconversion. Kidney biopsy and acute renal failure (ARF) was diagnosed. Urinalysis revealed acute tubular necrosis and abundant casts, consist- showed hypercellularity, with extensive tubular epithelial ing of bilirubin pigment. With conservative treatment, the cells, granular casts and amorphous material. A bone mar- patient fully recovered from cholemic nephrosis, an uncom- row aspirate ruled out haemophagocytic syndrome. A com- mon condition, not described after EBV infection before. puted tomography of the chest and abdomen showed bilateral pleural effusion, hepatomegaly (18 cm) and spleno- Keywords: acute kidney failure; cholemic nephrosis; Epstein-Barr virus; megaly (15 cm), but no hydronephrosis. Because of progres- Fouchet; hyperbilirubinaemia sive oliguric renal failure with fluid overload, intermittent haemodialysis was started. -

Renal Papillary Necrosis-A Sixteen-Year Clinical 1
Renal Papillary Necrosis-A Sixteen-Year Clinical 1 Matthew D. Griffin, Erik J. Bergstralh, and Timothy S. Larson2 emphasizes that continued recognition of papillary M.D. Griffin, IS. Larson. Division of Nephrology and necrosis will necessitate an awareness of its clinical Internal Medicine. Mayo Clinic and Mayo Foundation. spectrum in the context of current diagnostic and Rochester, MN therapeutic practice. E.J. Bergstralh. Section of Biostatistics, Mayo Clinic and Key Words: Diabetes mellitus. analgesic abuse, urinary tract Mayo Foundation. Rochester. MN infection, urinan, tract obstruction, sickle cell disease (J. Am. Soc. Nephrol. 1995; 6:248-256) R enal papillary necrosis (RPN) is a term used to describe a particular form of renal damage in ABSTRACT which any or all of the papillae undergo selective This study sought to characterize patients with renal necrosis in a manner that can be clearly demonstrated papillary necrosis seen at one tertiary referral center radiologically or histologically. The clinical syndromes by reviewing medical records of patients with a con- that have been described as accompanying this pro- cess include acute fulminant renal failure, obstruc- firmed diagnosis between January 1 , 1976 and Sep- tion by a sloughed papilla, hematuria, and loin pain, tember 1 , 1992. One hundred sixty-five cases were passage of tissue in the urine, recurrent or severe identified. The mean age at diagnosis was 57 yr (SD urinary tract infection (UT!), and chronic renal failure 15). The female-to-male ratio was 1 .1 :1 .0. Ninety-two (1-8). percent of patients were white. Seventy-seven percent From its first clear description by Friedreich in 1877 of cases were unsuspected before diagnosis, and in association with benign prostatic obstruction (9), 16% were diagnosed at autopsy. -
Histopathological Patterns of Nephrocalcinosis: a Phosphate Type Can Be Distinguished from a Calcium Type
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Nephrol Dial Transplant (2012) 27: 1122–1131 doi: 10.1093/ndt/gfr414 Advance Access publication 29 July 2011 Histopathological patterns of nephrocalcinosis: a phosphate type can be distinguished from a calcium type Thorsten Wiech1,2, Helmut Hopfer3, Ariana Gaspert4, Susanne Banyai-Falger5, Martin Hausberg6, Josef Schro¨der7, Martin Werner2 and Michael J. Mihatsch3 1Centre of Chronic Immunodeficiency, University Medical Center Freiburg, University of Freiburg, Freiburg, Germany, 2Institute of Pathology, University Hospital Freiburg, Freiburg, Germany, 3Institute for Pathology, University Hospital Basel, Basel, Switzerland, 4Institute of Surgical Pathology, University Hospital Zurich, Zurich, Switzerland, 5Klinik St Anna, Luzern, Switzerland, 6Sta¨dtisches Klinikum Karlsruhe, Karlsruhe, Germany and 7Institute of Pathology, University Hospital Regensburg, Regensburg, Germany Correspondence and offprint requests to: Thorsten Wiech; E-mail: [email protected] Abstract different chemical composition, such as calcium oxalate or Background. The etiology of nephrocalcinosis is variable. calcium phosphate, and can, but does not have to be associ- In this study, we wanted to elucidate whether the histopa- ated with nephrolithiasis. Pathologists distinguish between thological appearance of calcium phosphate deposits oxalosis and nephrocalcinosis, the latter again covering dif- provides information about possible etiology. ferent compositions but excluding calcium oxalate. Tradi- Methods. Autopsy cases from the years 1988 to 2007 and tionally, pathologists used the term ‘calcium nephrosis’ for native kidney biopsies from a 50-year period (1959–2008) dystrophic calcification i.e. calcification of necrosis and the with nephrocalcinosis were identified. The biopsy cases term ‘nephrocalcinosis’ for metastatic calcification [1]. -

Urinary Tract Disorders in Cattle
Urinary Tract Disorders in Cattle Thomas J. Divers, D. V.M New Bolton Center Kennett Square, PA Acute Renal Failure in Cattle a dehydrated animal (less than 1.022), and microscopic hematuria is usually present. Acute Tubular Necrosis Acute tubular necrosis is the most common patho Epidemiology: Acorn or oak bud poisoning is the most logic cause of acute renal failure (ARF) in cattle. This frequent in the autumn and spring. It often occurs in the results from renal ischemia or nephrotoxins. Renal autumn in association with sprouted acorns and the ischemia occurs most frequently in cattle with acute severe weaning of calves in a cow/calf operation onto a pasture of mastitis, septic metritis, abomasal torsion, salmonella, or sprouting acorns. Oak bud poisoning occurs in the spring other severe septic processes. The most common toxins and may be associated with late winter snows covering causing acute tubular necrosis in cattle are: other available grazing material. Aminoglycoside nephrotoxicity is most frequent in Quercus spp. (oak) dehydrated patients. Oxytetracycline nephrotoxicity is Halogeton glomerultos usually associated with overdosing of the drug, although it Sarcobatus vermiculatus (greasewood) may occur at normal doses in septic cattle. Oxalis spp. (soursob) Anaranthus spp. (pigwood) Treatment: Treatment is aimed at restoring intravascular Kochia spp. fluid volume by the administration of intravenous fluids Rumex (sorrel, dock) containing sodium chloride and calcium and potassium Chenopodium album (lambs quarter) when needed. The animal should be removed from the Rheum Rhaporticum (rhubarb) source of the toxin. Most cattle with acute tubular Salsola pestfiter (Russian thistle) nephrosis are polyuric, and if the disease process has not Lantana camara be present for several days, the prognosis is usually good if Isotropis the predisposing cause can be corrected. -

Experimental and Clinical Nephrosis J
Henry Ford Hospital Medical Journal Volume 9 | Number 2 Article 13 7-1961 Experimental And Clinical Nephrosis J. M. Bandera J. M. O'Neill Gordon Manson Follow this and additional works at: https://scholarlycommons.henryford.com/hfhmedjournal Part of the Life Sciences Commons, Medical Specialties Commons, and the Public Health Commons Recommended Citation Bandera, J. M.; O'Neill, J. M.; and Manson, Gordon (1961) "Experimental And Clinical Nephrosis," Henry Ford Hospital Medical Bulletin : Vol. 9 : No. 2 , 339-350. Available at: https://scholarlycommons.henryford.com/hfhmedjournal/vol9/iss2/13 This Article is brought to you for free and open access by Henry Ford Health System Scholarly Commons. It has been accepted for inclusion in Henry Ford Hospital Medical Journal by an authorized editor of Henry Ford Health System Scholarly Commons. For more information, please contact [email protected]. EXPERIMENTAL AND CLINICAL NEPHROSIS* J. M. BANDERA, M.D., J. M. O'NEILL, M.D., AND GORDON MANSON, M.D. DR. MANSON In our last basic science seminar, Drs. Caldwell, Neher and Hamilton Smith dealt with the ultrastructure of the nephron which constituted a fitting introduction to our consideration this afternoon of the experimental and clinical aspects of nephrosis. Nephrosis is a systemic disease with disturbances in many a.reas of metabolism. In the broad sense of the word, there are many causes of the clinical syndrome we call nephrosis. These have been reviewed at length by Adams^ in his excellent monograph. For the present seminar, however, we shall confine our dis cussion to consideration of a clinical entity characterized by proteinuria, hypopro teinemia and hyperlipemia.