Activation of the Renin-Angiotensin System and Chronic Hypoxia of the Kidney
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Signatures of Tissue-Specific
Developmental Cell Resource Molecular Signatures of Tissue-Specific Microvascular Endothelial Cell Heterogeneity in Organ Maintenance and Regeneration Daniel J. Nolan,1,6 Michael Ginsberg,1,6 Edo Israely,1 Brisa Palikuqi,1 Michael G. Poulos,1 Daylon James,1 Bi-Sen Ding,1 William Schachterle,1 Ying Liu,1 Zev Rosenwaks,2 Jason M. Butler,1 Jenny Xiang,4 Arash Rafii,1,7 Koji Shido,1 Sina Y. Rabbany,1,8 Olivier Elemento,3 and Shahin Rafii1,5,* 1Department of Genetic Medicine, Howard Hughes Medical Institute 2Ronald O. Perelman and Claudia Cohen Center for Reproductive Medicine 3HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine 4Genomics Resource Core Facility Weill Cornell Medical College, New York, NY 10065, USA 5Ansary Stem Cell Institute, New York, NY 10065, USA 6Angiocrine Bioscience, New York, NY 10065, USA 7Weill Cornell Medical College-Qatar, Stem Cell and Microenvironment Laboratory, Education City, Qatar Foundation, Doha 24144, Qatar 8Bioengineering Program, Hofstra University, Hempstead, NY 11549, USA *Correspondence: srafi[email protected] http://dx.doi.org/10.1016/j.devcel.2013.06.017 SUMMARY been appreciated. Capillary ECs of the blood brain barrier (BBB) form a restrictive environment for passage between the Microvascular endothelial cells (ECs) within different brain tissue and the circulating blood. Many of the trafficking pro- tissues are endowed with distinct but as yet unrecog- cesses that are passive in other vascular beds are tightly nized structural, phenotypic, and functional attri- controlled in the brain (Rubin and Staddon, 1999). As opposed butes. We devised EC purification, cultivation, to the BBB, the capillary ECs of the kidney glomeruli are fenes- profiling, and transplantation models that establish trated for the filtration of the blood (Churg and Grishman, tissue-specific molecular libraries of ECs devoid of 1975). -

Role of New Podocyte-Associated Proteins in the Renal Ultrafiltration Barrier
From the DEPARTMENT OF LABORATORY MEDICINE Karolinska Institutet, Stockholm, Sweden ROLE OF NEW PODOCYTE-ASSOCIATED PROTEINS IN THE RENAL ULTRAFILTRATION BARRIER Angelina Schwarz Stockholm 2019 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by Universitetsservice US-AB 2019. © Angelina Schwarz, 2019 ISBN 978-91-7831-452-2 Cover: confocal microscopy image of a mouse glomerulus (front) and electron microscopy image of a podocyte (back) Role of new podocyte-associated proteins in the renal ultrafiltration barrier THESIS FOR DOCTORAL DEGREE (Ph.D.) By Angelina Schwarz Principal Supervisor: Opponent: Assoc. Prof. Jaakko Patrakka, MD, PhD Prof. Rachel Lennon, MD, PhD Karolinska Institutet University of Manchester Department of Laboratory Medicine School of Biological Sciences Division of Pathology/ICMC Division of Cell Matrix and Regenerative Medicine Co-supervisor(s): Lwaki Ebarasi, PhD Examination Board: Karolinska Institutet Assoc. Prof. Sergiu-Bogdan Catrina, MD, PhD Department of Laboratory Medicine Karolinska Institutet Division of Pathology/ICMC Department of Molecular Medicine and Surgery Division of Growth and Metabolism Mark Lal, PhD AstraZeneca Prof. Bengt Fellström, MD, PhD Bioscience, Cardiovascular, Renal and Uppsala University Metabolism, Innovative Medicines Biotech Unit Department of Medical Sciences Division of Nephrology Assoc. Prof. Taija Mäkinen, PhD Uppsala University Department of Immunology, Genetics and Pathology Division of Vascular Biology ABSTRACT Chronic kidney disease (CKD) is a major health problem and an economical burden affecting people worldwide. The main causes of CKD are diabetes and hypertension and patient numbers keep increasing. In many cases, CKD is progressive leading to end stage renal disease (ESRD), a condition that can be treated only through chronic dialysis or renal transplantation. -

Ward's Renal Lobule Model
Ward’s Renal Lobule Model 470029-444 1. Arcuate artery and vein. 7. Descending thick limb of 12. Collecting tubule. 2. Interlobular artery and vein. Henle's loop. 13. Papillary duct of Bellini. 3. Afferent glomerular arteriole. 8. Thin segment of Henle's 14. Vasa recta. loop. 4. Efferent glomerular arteriole. 15. Capillary bed of cortex (extends 9. Ascending thick limb of through entire cortex). 5. Renal corpuscle (glomerulus Henle's loop. plus Bowman's capsule). 10. Distal convoluted tubule. 16. Capillary bed of medulla (extends 6. Proximal convoluted tubule. 11. Arched connecting tubule. through entire medulla). MANY more banks of glomeruli occur in the cortex than are represented on the model, and the proportionate length of the medullary elements has been greatly reduced. The fundamental physiological unit of the kidney is the nephron, consisting of the glomerulus, Bowman's capsule, the proximal convoluted tubule, Henle's loop, and the distal convoluted tubule. The blood is filtered in the glomerulus, water and soluble substances, except blood proteins, passing into Bowman's capsule in the same proportions as they occur in the blood. In the proximal tubule water and certain useful substances are resorbed from the provisional urine, while some further components may be added to it by secretory activity on the part of the tubular epithelium. In the remainder of the tubule, resorption of certain substances is continued, while the urine is concentrated further by withdrawal of water. The finished urine flows through the collecting tubules without further change. Various kinds of loops occur, varying in length of the thin segment, and in the level to which they descend into the medulla. -

Terminology Resource File
Terminology Resource File Version 2 July 2012 1 Terminology Resource File This resource file has been compiled and designed by the Northern Assistant Transfusion Practitioner group which was formed in 2008 and who later identified the need for such a file. This resource file is aimed at Assistant Transfusion Practitioners to help them understand the medical terminology and its relevance which they may encounter in the patient’s medical and nursing notes. The resource file will not include all medical complaints or illnesses but will incorporate those which will need to be considered and appreciated if a blood component was to be administered. The authors have taken great care to ensure that the information contained in this document is accurate and up to date. Authors: Jackie Cawthray Carron Fogg Julia Llewellyn Gillian McAnaney Lorna Panter Marsha Whittam Edited by: Denise Watson Document administrator: Janice Robertson ACKNOWLEDGMENTS We would like to acknowledge the following people for providing their valuable feedback on this first edition: Tony Davies Transfusion Liaison Practitioner Rose Gill Transfusion Practitioner Marie Green Transfusion Practitioner Tina Ivel Transfusion Practitioner Terry Perry Transfusion Specialist Janet Ryan Transfusion Practitioner Dr. Hazel Tinegate Consultant Haematologist Reviewed July 2012 Next review due July 2013 Version 2 July 2012 2 Contents Page no. Abbreviation list 6 Abdominal Aortic Aneurysm (AAA) 7 Acidosis 7 Activated Partial Thromboplastin Time (APTT) 7 Acquired Immune Deficiency Syndrome -

Lymphatic Tissue Engineering and Regeneration Laura Alderfer1, Alicia Wei1 and Donny Hanjaya-Putra1,2,3,4,5,6*
Alderfer et al. Journal of Biological Engineering (2018) 12:32 https://doi.org/10.1186/s13036-018-0122-7 REVIEW Open Access Lymphatic Tissue Engineering and Regeneration Laura Alderfer1, Alicia Wei1 and Donny Hanjaya-Putra1,2,3,4,5,6* Abstract The lymphatic system is a major circulatory system within the body, responsible for the transport of interstitial fluid, waste products, immune cells, and proteins. Compared to other physiological systems, the molecular mechanisms and underlying disease pathology largely remain to be understood which has hindered advancements in therapeutic options for lymphatic disorders. Dysfunction of the lymphatic system is associated with a wide range of disease phenotypes and has also been speculated as a route to rescue healthy phenotypes in areas including cardiovascular disease, metabolic syndrome, and neurological conditions. This review will discuss lymphatic system functions and structure, cell sources for regenerating lymphatic vessels, current approaches for engineering lymphatic vessels, and specific therapeutic areas that would benefit from advances in lymphatic tissue engineering and regeneration. Keywords: Lymphangiogenesis, Tissue Engineering, Disease Modeling, Wound Healing, Lymphedema, Stem Cells, Biomaterials, Interstitial Fluid, Regeneration I. Introduction to the Lymphatic System and its role Interstitial fluid (IF) is a plasma filtrate that is generated Function by transcapillary filtration and is governed by Starling The lymphatic system is nearly ubiquitous in the human forces, the net difference between hydrostatic and body, present in all tissues except the epidermis, cartil- osmotic pressures, at the microcirculatory level [9]. In age, eye lens, cornea, retina, and bone marrow [1, 2]. order to maintain fluid homeostasis, lymph formation in The main functions of the lymphatic system include the initial lymphatic vessels must be balanced by the net fluid homeostasis and interstitial fluid drainage, immune flux of plasma being filtered out [4]. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Lymph and Lymphatic Vessels
Cardiovascular System LYMPH AND LYMPHATIC VESSELS Venous system Arterial system Large veins Heart (capacitance vessels) Elastic arteries Large (conducting lymphatic vessels) vessels Lymph node Muscular arteries (distributing Lymphatic vessels) system Small veins (capacitance Arteriovenous vessels) anastomosis Lymphatic Sinusoid capillary Arterioles (resistance vessels) Postcapillary Terminal arteriole venule Metarteriole Thoroughfare Capillaries Precapillary sphincter channel (exchange vessels) Copyright © 2010 Pearson Education, Inc. Figure 19.2 Regional Internal jugular vein lymph nodes: Cervical nodes Entrance of right lymphatic duct into vein Entrance of thoracic duct into vein Axillary nodes Thoracic duct Cisterna chyli Aorta Inguinal nodes Lymphatic collecting vessels Drained by the right lymphatic duct Drained by the thoracic duct (a) General distribution of lymphatic collecting vessels and regional lymph nodes. Figure 20.2a Lymphatic System Outflow of fluid slightly exceeds return Consists of three parts 1. A network of lymphatic vessels carrying lymph 1. Transports fluid back to CV system 2. Lymph nodes 1. Filter the fluid within the vessels 3. Lymphoid organs 1. Participate in disease prevention Lymphatic System Functions 1. Returns interstitial fluid and leaked plasma proteins back to the blood 2. Disease surveillance 3. Lipid transport from intestine via lacteals Venous system Arterial system Heart Lymphatic system: Lymph duct Lymph trunk Lymph node Lymphatic collecting vessels, with valves Tissue fluid Blood Lymphatic capillaries Tissue cell capillary Blood Lymphatic capillaries capillaries (a) Structural relationship between a capillary bed of the blood vascular system and lymphatic capillaries. Filaments anchored to connective tissue Endothelial cell Flaplike minivalve Fibroblast in loose connective tissue (b) Lymphatic capillaries are blind-ended tubes in which adjacent endothelial cells overlap each other, forming flaplike minivalves. -

Introduction to Capillary Electrophoresis
Contents About this handbook..................................................................................... ii Acronyms and symbols used ....................................................................... iii Capillary electrophoresis ...............................................................................1 Electrophoresis terminology ..........................................................................3 Electroosmosis ...............................................................................................4 Flow dynamics, efficiency, and resolution ....................................................6 Capillary diameter and Joule heating ............................................................9 Effects of voltage and temperature ..............................................................11 Modes of capillary electrophoresis ..............................................................12 Capillary zone electrophoresis ..........................................................12 Isoelectric focusing ...........................................................................18 Capillary gel electrophoresis ............................................................21 Isotachophoresis ...............................................................................26 Micellar electrokinetic capillary chromatography ............................28 Selecting the mode of electrophoresis .........................................................36 Approaches to methods development by CZE and MECC .........................37 -
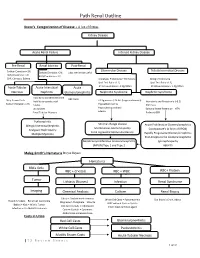
Path Renal Outline
Path Renal Outline Krane’s Categorization of Disease + A lot of Extras Kidney Disease Acute Renal Failure Intrinsic Kidney Disease Pre‐Renal Renal Intrinsic Post‐Renal Sodium Excretion <1% Glomerular Disease Tubulointerstitial Disease Sodium Excretion < 1% Sodium Excretion >2% Labs aren’t that useful BUN/Creatinine > 20 BUN/Creatinine < 10 CHF, Cirrhosis, Edema Urinalysis: Proteinuria + Hematuria Benign Proteinuria Spot Test Ratio >1.5, Spot Test Ratio <1.5, Acute Tubular Acute Interstitial Acute 24 Urine contains > 2.0g/24hrs 24 Urine contains < 1.0g/24hrs Necrosis Nephritis Glomerulonephritis Nephrotic Syndrome Nephritic Syndrome Inability to concentrate Urine RBC Casts Dirty Brown Casts Inability to secrete acid >3.5g protein / 24 hrs (huge proteinuria) Hematuria and Proteinuria (<3.5) Sodium Excretion >2% Edema Hypoalbuminemia RBC Casts Hypercholesterolemia Leukocytes Salt and Water Retention = HTN Focal Tubular Necrosis Edema Reduced GFR Pyelonephritis Minimal change disease Allergic Interstitial Nephritis Acute Proliferative Glomerulonephritis Membranous Glomerulopathy Analgesic Nephropathy Goodpasture’s (a form of RPGN) Focal segmental Glomerulosclerosis Rapidly Progressive Glomerulonephritis Multiple Myeloma Post‐Streptococcal Glomerulonephritis Membranoproliferative Glomerulonephritis IgA nephropathy (MPGN) Type 1 and Type 2 Alport’s Meleg‐Smith’s Hematuria Break Down Hematuria RBCs Only RBC + Crystals RBC + WBC RBC+ Protein Tumor Lithiasis (Stones) Infection Renal Syndrome Imaging Chemical Analysis Culture Renal Biopsy Calcium -

Management of the Nephrotic Syndrome GAVIN C
Arch Dis Child: first published as 10.1136/adc.43.228.257 on 1 April 1968. Downloaded from Personal Practice* Arch. Dis. Childh., 1968, 43, 257. Management of the Nephrotic Syndrome GAVIN C. ARNEIL From the Department of Child Health, University of Glasgow The nephrotic syndrome may be defined as gross (3) Idiopathic nephrosis. This may be re- proteinuria, predominantly albuminuria, with con- garded either as a primary disease, or as a secondary sequent selective hypoproteinaemia; usually accom- disorder, with the primary cause or causes still panied by oedema, ascites, and hyperlipaemia, and unknown. It is the common form of nephrosis in sometimes by systemic hypertension, azotaemia, or childhood, but the less common in adult patients. excessive erythrocyturia. Unless otherwise specified, the present discussion concerns idiopathic nephrosis. Grouping of Nephrotic Syndrome The syndrome falls into three groups. Assessment of the Case Full history and examination helps to exclude (1) Congenital nephrosis. A group of dis- many secondary forms of the disease. Blood orders present at and presenting soon after birth, pressure is recorded employing as broad a cuff as which are familial and may be acquired or inherited. practicable, and the width of the cuff used is Persistent oedema attracts attention in the first recorded as of importance in comparative estima- weeks of life. The disease may be rapidly lethal, tions. A careful search for concomitant infection but occasionally the patient will survive for a year is made, particularly to exclude pneumococcal copyright. or more without fatal infection or lethal azotaemia. peritonitis or low grade cellulitis. In a patient The disease pattern tends to run true within a previously treated elsewhere, evidence of steroid family. -

The Nephrotic Syndrome in Early Infancy: a Report of Three Cases* by H
Arch Dis Child: first published as 10.1136/adc.32.163.167 on 1 June 1957. Downloaded from THE NEPHROTIC SYNDROME IN EARLY INFANCY: A REPORT OF THREE CASES* BY H. McC. GILES, R. C. B. PUGH, E. M. DARMADY, FAY STRANACK and L. I. WOOLF From the Department of Paediatrics, St. Mary's Hospital, The Hospital for Sick Children, Great Ormond Street, London, and the Central Laboratory, Portsmouth (RECEIVED FOR PUBLICATION, DECEMBER 12, 1956) The nephrotic syndrome is characterized by evidence of genetic aetiology inasmuch as two of generalized oedema, hypoproteinaemia with diminu- the infants were brother and sister and both sets of tion or reversal of the albumin/globulin ratio, hyper- parents were cousins. Further points of interest lipaemia and gross albuminuria. Haematuria is not were the demonstration of anisotropic crystalline prominent; hypertension and azotaemia, except in material in alcohol-fixed tissues from all three the terminal stages, are slight and transient and infants, and the discovery on renal microdissection often do not occur at all. Recognition of this of lesions of the proximal tubule recalling those syndrome presents no difficulty but in many cases, found in Fanconi-Lignac disease (cystinosis). particularly in childhood, its cause remains obscure. Occasionally certain drugs such as troxidone or mercury, or certain diseases such as amyloidosis, Case Reports pyelonephritis, diabetes, disseminated lupus erythe- The parents of the first two patients (and the paternal copyright. matosus or renal vein thrombosis, can be in- grandparents) are first cousins. They, and a sister born criminated. In cases which develop renal failure three years before the first of her affected siblings, have and come to necropsy the lesions of glomerulo- been investigated in detail and show no sign of renal II disease. -

L25 Kidney2 to Post
Vert Phys PCB3743 Kidney 1 Fox Chapter 17 part 1 © T. Houpt, Ph.D. 1 Kidney Function Remove waste chemicals, while reabsorbing nutrients 1. Filter plasma from blood (including water & water-soluble nutrients) 2. Reabsorb Na+ : essential to maintain high extracellular [Na+] 3. Reabsorb H20 : essential to maintain body fluid volume 4. Reabsorb glucose and other nutrients 5. Reabsorb HCO3 / secrete H+ to maintain pH 2 Toxicity of Ammonia (NH3) 1. NH3 -> NH4+ very basic 2. NH3 is metabolic poison • Fish allow NH3 to diffuse into surrounding water • Birds & Reptiles convert NH3 to uric acid, which is not water soluble and is excreted in the feces. • Mammals convert NH3 to urea (CH4N2O), which is non-toxic and water soluble for excretion by kidney 1828: first organic synthesis: the production of urea without living tissue. → AgNCO + NH4Cl (NH2)2CO + AgCl 3 Krebs Cycle http://hyperphysics.phy-astr.gsu.edu/hbase/biology/tca.html 4 Too much ammonia removes a-ketoglutarate from Krebs cycle, so starves cells of ATP http://www.ucl.ac.uk/~ucbcdab/urea/amtox.htm 5 Urea - waste product of excess amino acid metabolism amino acid metabolism toxic! 2 liver ammonia + CO 6 Chapter 17: Anatomy of the Kidney Kidney Function Filter excess and waste chemicals (water soluble) from the blood. (excess water, Na+, urea, glucose > 200 mg/100ml) Kidney Structures cortex (bark): reddish brown, lots of capillaries medulla (inner region): striped with capillaries & collecting ducts; divided into renal pyramids urine -> collecting ducts -> minor calyces -> major calyces calyx; calyces -> renal pelvis-> ureters -> urinary bladder -> urethra “cup” high surface area for exchange, then to storage and outside ren- Latin for kidney nephro- Greek for kidney -uria - problem with urine, e.g.