Analysis of the Role of Purα in the Pathogenesis of Alzheimer's Disease Based on RNA-Seq and Chip-Seq
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Intrinsically Disordered Proteins of Myelin in Health and Disease
cells Review Flexible Players within the Sheaths: The Intrinsically Disordered Proteins of Myelin in Health and Disease Arne Raasakka 1 and Petri Kursula 1,2,* 1 Department of Biomedicine, University of Bergen, Jonas Lies vei 91, NO-5009 Bergen, Norway; [email protected] 2 Faculty of Biochemistry and Molecular Medicine & Biocenter Oulu, University of Oulu, Aapistie 7A, FI-90220 Oulu, Finland * Correspondence: [email protected] Received: 30 January 2020; Accepted: 16 February 2020; Published: 18 February 2020 Abstract: Myelin ensheathes selected axonal segments within the nervous system, resulting primarily in nerve impulse acceleration, as well as mechanical and trophic support for neurons. In the central and peripheral nervous systems, various proteins that contribute to the formation and stability of myelin are present, which also harbor pathophysiological roles in myelin disease. Many myelin proteins have common attributes, including small size, hydrophobic segments, multifunctionality, longevity, and regions of intrinsic disorder. With recent advances in protein biophysical characterization and bioinformatics, it has become evident that intrinsically disordered proteins (IDPs) are abundant in myelin, and their flexible nature enables multifunctionality. Here, we review known myelin IDPs, their conservation, molecular characteristics and functions, and their disease relevance, along with open questions and speculations. We place emphasis on classifying the molecular details of IDPs in myelin, and we correlate these with their various functions, including susceptibility to post-translational modifications, function in protein–protein and protein–membrane interactions, as well as their role as extended entropic chains. We discuss how myelin pathology can relate to IDPs and which molecular factors are potentially involved. Keywords: myelin; intrinsically disordered protein; multiple sclerosis; peripheral neuropathies; myelination; protein folding; protein–membrane interaction; protein–protein interaction 1. -

How Does Protein Zero Assemble Compact Myelin?
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 13 May 2020 doi:10.20944/preprints202005.0222.v1 Peer-reviewed version available at Cells 2020, 9, 1832; doi:10.3390/cells9081832 Perspective How Does Protein Zero Assemble Compact Myelin? Arne Raasakka 1,* and Petri Kursula 1,2 1 Department of Biomedicine, University of Bergen, Jonas Lies vei 91, NO-5009 Bergen, Norway 2 Faculty of Biochemistry and Molecular Medicine & Biocenter Oulu, University of Oulu, Aapistie 7A, FI-90220 Oulu, Finland; [email protected] * Correspondence: [email protected] Abstract: Myelin protein zero (P0), a type I transmembrane protein, is the most abundant protein in peripheral nervous system (PNS) myelin – the lipid-rich, periodic structure that concentrically encloses long axonal segments. Schwann cells, the myelinating glia of the PNS, express P0 throughout their development until the formation of mature myelin. In the intramyelinic compartment, the immunoglobulin-like domain of P0 bridges apposing membranes together via homophilic adhesion, forming a dense, macroscopic ultrastructure known as the intraperiod line. The C-terminal tail of P0 adheres apposing membranes together in the narrow cytoplasmic compartment of compact myelin, much like myelin basic protein (MBP). In mouse models, the absence of P0, unlike that of MBP or P2, severely disturbs the formation of myelin. Therefore, P0 is the executive molecule of PNS myelin maturation. How and when is P0 trafficked and modified to enable myelin compaction, and how disease mutations that give rise to incurable peripheral neuropathies alter the function of P0, are currently open questions. The potential mechanisms of P0 function in myelination are discussed, providing a foundation for the understanding of mature myelin development and how it derails in peripheral neuropathies. -

Pathological Relationships Involving Iron and Myelin May Constitute a Shared Mechanism Linking Various Rare and Common Brain Diseases
Rare Diseases ISSN: (Print) 2167-5511 (Online) Journal homepage: http://www.tandfonline.com/loi/krad20 Pathological relationships involving iron and myelin may constitute a shared mechanism linking various rare and common brain diseases Moones Heidari, Sam H. Gerami, Brianna Bassett, Ross M. Graham, Anita C.G. Chua, Ritambhara Aryal, Michael J. House, Joanna F. Collingwood, Conceição Bettencourt, Henry Houlden, Mina Ryten , John K. Olynyk, Debbie Trinder, Daniel M. Johnstone & Elizabeth A. Milward To cite this article: Moones Heidari, Sam H. Gerami, Brianna Bassett, Ross M. Graham, Anita C.G. Chua, Ritambhara Aryal, Michael J. House, Joanna F. Collingwood, Conceição Bettencourt, Henry Houlden, Mina Ryten , John K. Olynyk, Debbie Trinder, Daniel M. Johnstone & Elizabeth A. Milward (2016) Pathological relationships involving iron and myelin may constitute a shared mechanism linking various rare and common brain diseases, Rare Diseases, 4:1, e1198458, DOI: 10.1080/21675511.2016.1198458 To link to this article: http://dx.doi.org/10.1080/21675511.2016.1198458 © 2016 The Author(s). Published with View supplementary material license by Taylor & Francis Group, LLC© Moones Heidari, Sam H. Gerami, Brianna Bassett, Ross M. Graham, Anita C.G. Chua, Ritambhara Aryal, Michael J. House, Joanna Accepted author version posted online: 22 Submit your article to this journal JunF. Collingwood, 2016. Conceição Bettencourt, PublishedHenry Houlden, online: Mina 22 Jun Ryten, 2016. for the UK Brain Expression Consortium (UKBEC), John K. Olynyk, Debbie Trinder, Daniel M. Johnstone,Article views: and 541 Elizabeth A. Milward. View related articles View Crossmark data Citing articles: 2 View citing articles Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=krad20 Download by: [University of Newcastle, Australia] Date: 17 May 2017, At: 19:57 RARE DISEASES 2016, VOL. -
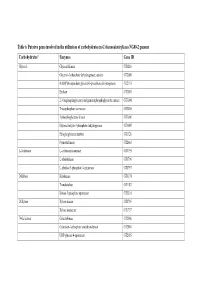
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

An Investigation on Catalysis of Acylaminoacyl Peptidases
An investigation on catalysis of acylaminoacyl peptidases PhD Thesis András László Kiss Doctorate School of Biology School leader: Prof. Anna Erdei Structural Biochemistry Programme Programme leader: Prof. László Gráf Supervisor: Prof. László Polgár Institute of Enzymology, Biological Research Center, Hungarian Academy of Sciences Budapest 2007 1. Introduction Serine peptidases contain two residues at the active site in addition to the catalytic triad (His, Asp, Ser), which form a cavity called “oxyanion hole” that accommodates the negatively charged oxyanion in the transition state of the catalysis and donate two H- bonds. Enzymes of the prolyl oligopeptidase (POP) family (acylaminoacyl peptidase, prolyl oligopeptidase, dipeptidyl peptidase IV and oligopeptidase B) are extensively studied. These enzymes are larger than classical serine peptidases and are composed of a peptidase domain with an α/β hydrolase fold and a β-propeller domain. POP and oligopeptidase B are monomeric enzymes and endopeptidases, while the exopeptidase acylaminoacyl peptidase and dipeptidyl peptidase IV are tetrameric and dimeric enzymes, respectively. Acylaminoacyl peptidase (AAP) cleaves acylated amino acids from the N-terminus of the N-acylated peptides that plays important role in many biological and disease processes. Human AAP is encoded by the DNF15S2 locus on the short arm of chromosome 3 at the region 21, which suffers deletions in small cell lung carcinomas, and renal carcinomas, resulting in deficiency in the expression of the enzyme. Acylaminoacyl peptidase is also supposed to be involved in the degradation of oxidatively damaged proteins in cells and can be associated with various diseases where damaged proteins aggregate. Its involvement in cataract formation and in the breakdown of immunogenic formylmethionyl-peptides in the digestive tract after bacterial attack was also suggested. -

High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma
molecules Article High-Resolution Mass Spectrometry-Based Approaches for the Detection and Quantification of Peptidase Activity in Plasma Elisa Maffioli 1,2 , Zhenze Jiang 3, Simona Nonnis 1,2 , Armando Negri 1,2, Valentina Romeo 1, Christopher B. Lietz 3, Vivian Hook 3,4, Giuseppe Ristagno 5, Giuseppe Baselli 6, Erik B. Kistler 7,8 , Federico Aletti 9, Anthony J. O’Donoghue 3,* and Gabriella Tedeschi 1,2,* 1 Department of Veterinary Medicine, University of Milano, 20133 Milano, Italy; elisa.maffi[email protected] (E.M.); [email protected] (S.N.); [email protected] (A.N.); [email protected] (V.R.) 2 Centre for Nanostructured Materials and Interfaces (CIMAINA), University of Milano, 20133 Milano, Italy 3 Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego, La Jolla, CA 92093, USA; [email protected] (Z.J.); [email protected] (C.B.L.); [email protected] (V.H.) 4 Department of Neurosciences, School of Medicine, University of California San Diego, La Jolla, CA 92093, USA 5 Department of Pathophysiology and Transplantation, University of Milan, 20133 Milan, Italy; [email protected] 6 Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano, 20133 Milan, Italy; [email protected] 7 Department of Anesthesiology & Critical Care, University of California San Diego, La Jolla, CA 92093, USA; [email protected] 8 Department of Anesthesiology & Critical Care, VA San Diego HealthCare System, San Diego, CA 92161, USA 9 Department of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA; [email protected] * Correspondence: [email protected] (A.J.O.); [email protected] (G.T.); Tel.: +1-8585345360 (A.J.O.); +39-02-50318127 (G.T.) Academic Editor: Paolo Iadarola Received: 28 July 2020; Accepted: 4 September 2020; Published: 6 September 2020 Abstract: Proteomic technologies have identified 234 peptidases in plasma but little quantitative information about the proteolytic activity has been uncovered. -

Myelination Is Delayed During Postnatal Brain Development in the Mdx Mouse Model of Duchenne Muscular Dystrophy Azeez Aranmolate, Nathaniel Tse and Holly Colognato*
Aranmolate et al. BMC Neurosci (2017) 18:63 DOI 10.1186/s12868-017-0381-0 BMC Neuroscience RESEARCH ARTICLE Open Access Myelination is delayed during postnatal brain development in the mdx mouse model of Duchenne muscular dystrophy Azeez Aranmolate, Nathaniel Tse and Holly Colognato* Abstract Background: In Duchenne muscular dystrophy (DMD), the loss of the dystrophin component of the dystrophin- glycoprotein complex (DGC) compromises plasma membrane integrity in skeletal muscle, resulting in extensive muscle degeneration. In addition, many DMD patients exhibit brain defcits in which the cellular etiology remains poorly understood. We recently found that dystroglycan, a receptor component of the DGC that binds intracellularly to dystrophin, regulates the development of oligodendrocytes, the myelinating glial cells of the brain. Results: We investigated whether dystrophin contributes to oligodendroglial function and brain myelination. We found that oligodendrocytes express up to three dystrophin isoforms, in conjunction with classic DGC components, which are developmentally regulated during diferentiation and in response to extracellular matrix engagement. We found that mdx mice, a model of DMD lacking expression of the largest dystrophin isoform, have delayed myelination and inappropriate oligodendrocyte progenitor proliferation in the cerebral cortex. When we prevented the expression of all oligodendroglial dystrophin isoforms in cultured oligodendrocytes using RNA interference, we found that later stages of oligodendrocyte maturation -

Can the Fact That Myelin Proteins Are Old and Break Down Explain the Origin of Multiple Sclerosis in Some People?
Journal of Clinical Medicine Review Can the Fact That Myelin Proteins Are Old and Break down Explain the Origin of Multiple Sclerosis in Some People? Roger J. W. Truscott * and Michael G. Friedrich Illawarra Health and Medical Research Institute, University of Wollongong, Wollongong, NSW 2522, Australia; [email protected] * Correspondence: [email protected]; Tel.: +61-2-4298-3503; Fax: +61-2-4221-8130 Received: 11 July 2018; Accepted: 12 September 2018; Published: 14 September 2018 Abstract: Recent discoveries may change the way that multiple sclerosis (MS) is viewed, particularly with regard to the reasons for the untoward immune response. The fact that myelin proteins are long-lived, and that by the time we are adults, they are extensively degraded, alters our perspective on the reasons for the onset of autoimmunity and the origin of MS. For example, myelin basic protein (MBP) from every human brain past the age of 20 years, is so greatly modified, that it is effectively a different protein from the one that was laid down in childhood. Since only a subset of people with such degraded MBP develop MS, a focus on understanding the mechanism of immune responses to central nervous system (CNS) antigens and cerebral immune tolerance appear to be worthwhile avenues to explore. In accord with this, it will be productive to examine why all people, whose brains contain large quantities of a “foreign antigen”, do not develop MS. Importantly for the potential causation of MS, MBP from MS patients breaks down differently from the MBP in aged controls. If the novel structures formed in these MS-specific regions are particularly antigenic, it could help explain the origin of MS. -

And Exopeptidases in a Processing Enzyme System: Activation
Proc. Nail. Acad. Sci. USA Vol. 85, pp. 5468-5472, August 1988 Biochemistry Relationship between endo- and exopeptidases in a processing enzyme system: Activation of an endoprotease by the aminopeptidase B-like activity in somatostatin-28 convertase (brain cortex/basic amino acid pairs/peptide substrates/protease inhibitors/prohormone maturation) SOPHIE GOMEZ, PABLO GLUSCHANKOF, AGNES LEPAGE, AND PAUL COHEN Groupe de Neurobiochimie Cellulaire et Moldculaire, Universitd Pierre et Marie Curie, Unitd Associde 554 au Centre National de la Recherche Scientifique, 96 boulevard Raspail, 75006 Paris, France Communicated by I. Robert Lehman, April 8, 1988 (receivedfor review December 15, 1987) ABSTRACT The somatostatin-28 convertase activity in- somatostatin-14 and the amino-terminal dodecapeptide so- volved in vitro in the processing of somatostatin-28 into the matostatin-28-(1-12) (16). neuropeptides somatostatin-28-(1-12) and somatostatin-14 is We have described an endoprotease that cleaves the composed of an endoprotease and a basic aminopeptidase. We peptide bond on the amino side ofthe Arg-Lys doublet in the report herein on the purification to apparent homogeneity of somatostatin-28 sequence (17, 18), releasing the somato- these two constituents and on their functional interrelationship. statin-28-(1-12) fragment and [Arg-2,Lys-1]somatostatin-14. In particular we observed that after various physicochemical The released [Arg-2,Lys-']somatostatin-14 is further proc- treatments, the 90-kDa endoprotease activity was recovered essed by an aminopeptidase B-like activity (18, 19) that is also both at this molecular mass and as a 45-kDa entity. Moreover, present in the preparation. We report herein the purification the production of [Arg2,LysJllsomatostatin-14 from somato- to apparent homogeneity ofthese two activities. -

Proteases for Biocatalysis
Proteases for biocatalysis for smarter chemical synthesis Biocatalysis Biocatalysis involves the implementation of natural catalysts, such as enzymes, in place of chemical catalysts in synthetic processes. Compared to chemical catalysts, enzymes offer: • higher reaction rates • milder reaction conditions • high reaction specificity with no side products This change can enable new, more sustainable routes for the production of intermediates and active pharmaceutical ingredients (APIs). However, please note Novozymes products do not comply with manufacturing according to pharmaceutical standards and Novozymes products must not be used as active pharmaceutical ingredients (APIs) or excipients. Biocatalysis has become an increasingly important tool for medicinal, process and polymer chemists, allowing the development of efficient and highly attractive synthetic processes on an industrial scale. Use of enzymes in catalysis is a well-established technology within the chemical industry. An advantage of enzymes in organic synthesis is their remarkable selective properties, which provide commercial benefits including: • high selectivity in production of single stereoisomers • fewer side reactions • less reprocessing and purification steps • easier product separation • less pollution The combination of all of these advantages leads to a reduction in costs. Enzyme catalysts work by lowering the activation energy (Ea‡) for a reaction, thus dramatically increasing the rate of the reaction. As a result, products are formed faster and reactions reach their equilibrium state more rapidly. Most enzyme reaction rates are millions of times faster than those of comparable uncatalyzed reactions. As with all catalysts, enzymes are not consumed by the reactions they catalyze, nor do they alter the equilibrium of these reactions. However, enzymes do differ from most other catalysts in that they are highly specific for their substrates. -

Chapter 11 Cysteine Proteases
CHAPTER 11 CYSTEINE PROTEASES ZBIGNIEW GRZONKA, FRANCISZEK KASPRZYKOWSKI AND WIESŁAW WICZK∗ Faculty of Chemistry, University of Gdansk,´ Poland ∗[email protected] 1. INTRODUCTION Cysteine proteases (CPs) are present in all living organisms. More than twenty families of cysteine proteases have been described (Barrett, 1994) many of which (e.g. papain, bromelain, ficain , animal cathepsins) are of industrial impor- tance. Recently, cysteine proteases, in particular lysosomal cathepsins, have attracted the interest of the pharmaceutical industry (Leung-Toung et al., 2002). Cathepsins are promising drug targets for many diseases such as osteoporosis, rheumatoid arthritis, arteriosclerosis, cancer, and inflammatory and autoimmune diseases. Caspases, another group of CPs, are important elements of the apoptotic machinery that regulates programmed cell death (Denault and Salvesen, 2002). Comprehensive information on CPs can be found in many excellent books and reviews (Barrett et al., 1998; Bordusa, 2002; Drauz and Waldmann, 2002; Lecaille et al., 2002; McGrath, 1999; Otto and Schirmeister, 1997). 2. STRUCTURE AND FUNCTION 2.1. Classification and Evolution Cysteine proteases (EC.3.4.22) are proteins of molecular mass about 21-30 kDa. They catalyse the hydrolysis of peptide, amide, ester, thiol ester and thiono ester bonds. The CP family can be subdivided into exopeptidases (e.g. cathepsin X, carboxypeptidase B) and endopeptidases (papain, bromelain, ficain, cathepsins). Exopeptidases cleave the peptide bond proximal to the amino or carboxy termini of the substrate, whereas endopeptidases cleave peptide bonds distant from the N- or C-termini. Cysteine proteases are divided into five clans: CA (papain-like enzymes), 181 J. Polaina and A.P. MacCabe (eds.), Industrial Enzymes, 181–195. -

LRP1 Regulates Peroxisome Biogenesis and Cholesterol
RESEARCH ARTICLE LRP1 regulates peroxisome biogenesis and cholesterol homeostasis in oligodendrocytes and is required for proper CNS myelin development and repair Jing-Ping Lin1, Yevgeniya A Mironova2, Peter Shrager3, Roman J Giger1,2,4,5* 1Department of Cell and Developmental Biology, University of Michigan School of Medicine, Ann Arbor, MI, United States; 2Cellular and Molecular Biology Graduate Program, University of Michigan Medical School, Ann Arbor, MI, United States; 3Department of Neuroscience, University of Rochester Medical Center, Rochester, NY, United States ; 4Department of Neurology, University of Michigan Medical School, Ann Arbor, MI, United States; 5Interdepartmental Neuroscience Graduate Program, University of Michigan Medical School, Ann Arbor, MI, United States Abstract Low-density lipoprotein receptor-related protein-1 (LRP1) is a large endocytic and signaling molecule broadly expressed by neurons and glia. In adult mice, global inducible (Lrp1flox/ flox;CAG-CreER) or oligodendrocyte (OL)-lineage specific ablation (Lrp1flox/flox;Pdgfra-CreER) of Lrp1 attenuates repair of damaged white matter. In oligodendrocyte progenitor cells (OPCs), Lrp1 is required for cholesterol homeostasis and differentiation into mature OLs. Lrp1-deficient OPC/ OLs show a strong increase in the sterol-regulatory element-binding protein-2 yet are unable to maintain normal cholesterol levels, suggesting more global metabolic deficits. Mechanistic studies revealed a decrease in peroxisomal biogenesis factor-2 and fewer peroxisomes in OL processes. / *For correspondence: Treatment of Lrp1À À OPCs with cholesterol or activation of peroxisome proliferator-activated [email protected] receptor-g with pioglitazone alone is not sufficient to promote differentiation; however, when combined, cholesterol and pioglitazone enhance OPC differentiation into mature OLs. Collectively, Competing interests: The our studies reveal a novel role for Lrp1 in peroxisome biogenesis, lipid homeostasis, and OPC authors declare that no competing interests exist.