TESIS DOCTORAL Spatio-Temporal Genetic Dynamics of European Hake
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

DEEP SEA LEBANON RESULTS of the 2016 EXPEDITION EXPLORING SUBMARINE CANYONS Towards Deep-Sea Conservation in Lebanon Project
DEEP SEA LEBANON RESULTS OF THE 2016 EXPEDITION EXPLORING SUBMARINE CANYONS Towards Deep-Sea Conservation in Lebanon Project March 2018 DEEP SEA LEBANON RESULTS OF THE 2016 EXPEDITION EXPLORING SUBMARINE CANYONS Towards Deep-Sea Conservation in Lebanon Project Citation: Aguilar, R., García, S., Perry, A.L., Alvarez, H., Blanco, J., Bitar, G. 2018. 2016 Deep-sea Lebanon Expedition: Exploring Submarine Canyons. Oceana, Madrid. 94 p. DOI: 10.31230/osf.io/34cb9 Based on an official request from Lebanon’s Ministry of Environment back in 2013, Oceana has planned and carried out an expedition to survey Lebanese deep-sea canyons and escarpments. Cover: Cerianthus membranaceus © OCEANA All photos are © OCEANA Index 06 Introduction 11 Methods 16 Results 44 Areas 12 Rov surveys 16 Habitat types 44 Tarablus/Batroun 14 Infaunal surveys 16 Coralligenous habitat 44 Jounieh 14 Oceanographic and rhodolith/maërl 45 St. George beds measurements 46 Beirut 19 Sandy bottoms 15 Data analyses 46 Sayniq 15 Collaborations 20 Sandy-muddy bottoms 20 Rocky bottoms 22 Canyon heads 22 Bathyal muds 24 Species 27 Fishes 29 Crustaceans 30 Echinoderms 31 Cnidarians 36 Sponges 38 Molluscs 40 Bryozoans 40 Brachiopods 42 Tunicates 42 Annelids 42 Foraminifera 42 Algae | Deep sea Lebanon OCEANA 47 Human 50 Discussion and 68 Annex 1 85 Annex 2 impacts conclusions 68 Table A1. List of 85 Methodology for 47 Marine litter 51 Main expedition species identified assesing relative 49 Fisheries findings 84 Table A2. List conservation interest of 49 Other observations 52 Key community of threatened types and their species identified survey areas ecological importanc 84 Figure A1. -

Trophic Relationships of Hake (Merluccius Capensis and M
SoV 2.10 Trophic Relationships of Hake (Merluccius capensis and M. paradoxus) and Sharks (Centrophorus squamosus, Deania calcea and D. profundorum) in the Northern (Namibia) Benguela Current region Author(s): Johannes A Iitembu and Nicole B Richoux Source: African Zoology, 50(4):273-279. Published By: Zoological Society of Southern Africa URL: http://www.bioone.org/doi/full/10.1080/15627020.2015.1079142 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. Usage of BioOne content is strictly limited to personal, educational, and non-commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. African Zoology 2015, 50(4): 273–279 Copyright © Zoological Society Printed in South Africa — All rights reserved of Southern Africa AFRICAN ZOOLOGY This is the final version of the article that is ISSN 1562-7020 EISSN 2224-073X published ahead of the print and online issue http://dx.doi.org/10.1080/15627020.2015.1079142 Trophic relationships of hake (Merluccius capensis and M. paradoxus) and sharks (Centrophorus squamosus, Deania calcea and D. -

Integrative Taxonomy, a New Tool for Fisheries Conservation
W&M ScholarWorks Presentations 10-9-2015 Integrative Taxonomy, a New Tool for Fisheries Conservation Adela Roa-Varon Virginia Institute of Marine Science Eric J. Hilton Virginia Institute of Marine Science Follow this and additional works at: https://scholarworks.wm.edu/presentations Part of the Aquaculture and Fisheries Commons, Biodiversity Commons, and the Zoology Commons Recommended Citation Roa-Varon, Adela and Hilton, Eric J.. "Integrative Taxonomy, a New Tool for Fisheries Conservation". 10-9-2015. VIMS 75th Anniversary Alumni Research Symposium. This Presentation is brought to you for free and open access by W&M ScholarWorks. It has been accepted for inclusion in Presentations by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Integrave Taxonomy: a New Tool for Fisheries Conservaon Adela Roa-Varon and Eric Hilton Virginia InsFtute of Marine Science, William & Mary College [email protected], [email protected] What is Integrave Taxonomy? The Process of Integrave Results to Date Glossary Taxonomy Baits libraries were designed based on Species play a central role in nearly all candidate single-copy markers for disciplines of biology. Therefore delimitaon Gadus morhua genome, using the Species are the fundamental of species has broad implicaons for efforts taxonomic unit for a wide array of MYBaits target enrichment system. A biological studies and applied fields ranging from biological conservaon to preliminary analysis was run to test the such as conservaon planning. comparave evoluFonary analyses. The rise of baits using Merluccius bilinearis and new genomic and bioinformacs tools led for Gadus morhua as a posiFve control. Species delimitaon: the process of species delimitaon is becoming increasingly Approximately, 1500 to 3000 target idenFfying species-level biological objecFve and integrave. -

Synopsis Iconographique Des Otolithes De Quelques Espèces De Poissons Des Côtes Ouest Africaines
Synopsis iconographique des otolithes de quelques espèces de poissons des côtes ouest africaines Jan Veen et Kristiaan Hoedemakers VEDA consultancy Synopsis iconographique des otolithes de quelques espèces de poissons des côtes ouest africaines Jan Veen1, Kristiaan Hoedemakers2 1. VEDA consultancy, Wieselseweg 110, 7345 CC Wenum Wiesel, The Netherlands 2. Kristiaan Hoedemakers, Minervastraat 23, 2640 Mortsel, Belgium Wetlands International 2005 Copyright 2005 Wetlands International ISBN 9058829553 Cette publication doit être citée comme suit: Veen, J., Hoedemakers, K., 2005, Synopsis iconographique des otolithes de quelques espèces de poissons des côtes ouest africaines. Wageningen, The Netherlands. Publié par Wetlands International www.wetlands.org Dessins: Kristiaan Hoedemakers et Dirk Nolf. Tous droits réservés. Photos: Cindy van Damme, Alterra. Tous droits réservés. Texte: Jan Veen et Kristiaan Hoedemakers Lay-out: Kristiaan Hoedemakers Les données et désignations géographiques employées dans ce rapport n’impliquent en aucune manière une expression quelconque de l’opinion de la part de Wetlands International sur le statut légal d’un pays quel qu’il soit, d’une région ou d’un territoire, ou concernant la délimitation de ses limites ou frontières. Synopsis iconographique des otolithes de quelques espèces de poissons des côtes ouest africaines Organismes d’appui et de collaboration VEDA consultancy - research, advice and training in ecology and geography, The Netherlands VEDA consultancy Directorate General for International Co-operation, Ministry of Foreign Affairs, The Netherlands Directorate for Nature Management, Ministry of Agriculture, Nature and Food Quality, The Netherlands Financé par le Ministère de l’Agriculture, de la Nature et de la Qualité de l’Alimentation et le Ministère des Affaires Etrangères des Pays-Bas, dans le cadre du Programme Biodiversité de la Politique Internationale 2002-2006 des Pays-Bas. -

Intrinsic Vulnerability in the Global Fish Catch
The following appendix accompanies the article Intrinsic vulnerability in the global fish catch William W. L. Cheung1,*, Reg Watson1, Telmo Morato1,2, Tony J. Pitcher1, Daniel Pauly1 1Fisheries Centre, The University of British Columbia, Aquatic Ecosystems Research Laboratory (AERL), 2202 Main Mall, Vancouver, British Columbia V6T 1Z4, Canada 2Departamento de Oceanografia e Pescas, Universidade dos Açores, 9901-862 Horta, Portugal *Email: [email protected] Marine Ecology Progress Series 333:1–12 (2007) Appendix 1. Intrinsic vulnerability index of fish taxa represented in the global catch, based on the Sea Around Us database (www.seaaroundus.org) Taxonomic Intrinsic level Taxon Common name vulnerability Family Pristidae Sawfishes 88 Squatinidae Angel sharks 80 Anarhichadidae Wolffishes 78 Carcharhinidae Requiem sharks 77 Sphyrnidae Hammerhead, bonnethead, scoophead shark 77 Macrouridae Grenadiers or rattails 75 Rajidae Skates 72 Alepocephalidae Slickheads 71 Lophiidae Goosefishes 70 Torpedinidae Electric rays 68 Belonidae Needlefishes 67 Emmelichthyidae Rovers 66 Nototheniidae Cod icefishes 65 Ophidiidae Cusk-eels 65 Trachichthyidae Slimeheads 64 Channichthyidae Crocodile icefishes 63 Myliobatidae Eagle and manta rays 63 Squalidae Dogfish sharks 62 Congridae Conger and garden eels 60 Serranidae Sea basses: groupers and fairy basslets 60 Exocoetidae Flyingfishes 59 Malacanthidae Tilefishes 58 Scorpaenidae Scorpionfishes or rockfishes 58 Polynemidae Threadfins 56 Triakidae Houndsharks 56 Istiophoridae Billfishes 55 Petromyzontidae -

Phylogenetic Prospecting for Cryptic Species of the Genus Merluccius (Actinopterygii: Merlucciidae)
Phylogenetic prospecting for cryptic species of the genus Merluccius (Actinopterygii: Merlucciidae) Montse Pérez1, María Fernández-Míguez1,2, Jesús Matallanas3, Domingo Lloris4 and Pablo Presa2,* 1AquaCOV, Centro Oceanográfico de Vigo, Instituto Español de Oceanografía, 36390 Vigo, Spain. 2CIM-Universidad de Vigo, Laboratorio de Recursos Genéticos Marinos, Facultad de Biología, 36310 Vigo, Spain. 3Unidad de Zoología, Departamento de Biología Animal, Biología Vegetal y Ecología, Universidad Autónoma de Barcelona, 08193, Spain. 4Institut de Ciències del Mar (CMIMA-CSIC), Barcelona, 08003, Spain *[email protected] a b c Supplementary Figure S1. Principal Coordinates Analysis (PCoA) built after the molecular variation of ITS1 variants using GenAlEx v6.503 [79]; a) Old World (OW) hakes (orange symbols: M. merluccius, M. senegalensis, M. capensis, M. polli and M. paradoxus), New World (NW) hakes (yellow symbols: M. productus, M. gayi, M. angustimanus, M. australis, M. albidus, M. hubbsi and M. bilinearis), and Atlantic cod (black symbol, Gadus morhua); b) Atlantic NW hakes (green symbols: M. albidus and M. bilinearis), Pacific NW hakes (blue symbols: M. australis, M. angustimanus, M. productus c) PCoA built after the molecular variation of ITS1Nes variants from Atlantic NW hakes (green symbols, M. albidus, M. bilinearis), Pacific NW hakes (blue symbols: M. angustimanus, M. productus, M. gayi), Austral NW hakes (orange symbols, M. hubbsi and M. australis) and morphotypes (red circled blue symbols: M. tasmanicus, M. patagonicus and M. polylepis). The European hake (merl, Merluccius merluccius) is the outgroup. a b Supplementary Figure S2. Phylogenetic reconstruction on ITS1 variants from genus Merluccius spp. using the substitution model HKY85+I+G. a) ML (-lnL = 1285.511) performed with PAUP v4.0 [84]). -

Tfm Hanane El Yaagoubi
Máster Internacional en GESTIÓN PESQUERA SOSTENIBLE (7ª edición: 2017-2019) TESIS presentada y públicamente defendida para la obtención del título de MASTER OF SCIENCE HANANE EL YAAGOUBI Septiembre 2019 MASTERENGESTIÓNPESQUERASOSTENIBLE (7ªedición: 2017-2019) Spatiotemporal variation of fishery patterns, demographic indices and spatial distribution of European hake, Merluccius merluccius, in the GSA 01 and GSA03 Hanane EL YAAGOUBI TESIS PRESENTADA Y PUBLICAMENTE DEFENDIDA PARA LA OBTENCIÓN DEL TÍTULO DE MASTER OF SCIENCE EN GESTIÓN PESQUERA SOSTENIBLE Alicante a…09.de Septiembre de2019 ii Spatiotemporal variation of fishery patterns, demographic indices and spatial distribution of European hake, Merluccius merluccius, in the GSA 01 and GSA03 Hanane EL YAAGOUBI Trabajo realizado en el Centro Oceanográfico de Baleares (COB) del Instituto Español de Oceanografía (IEO), España, bajo la dirección del Dr.Manuel HIDALGO y la Dra. Pilar Hernández Y presentado como requisito parcial para la obtención del Diploma Master of Science en Gestión Pesquera Sostenible otorgado por la Universidad de Alicante a través de Facultad de Ciencias y el Centro Internacional de Altos Estudios Agronómicos Mediterráneos (CIHEAM) a través del Instituto Agronómico Mediterráneo de Zaragoza(IAMZ). V B Tutor y Tutora Autora Fdo:Dr.Manuel Hidalgo y Dra. Pilar Hernández... Fdo: Hanane El yaagoubi................. Alicante ,a 25 de Septiembre 2019 iii iv Spatiotemporal variation of fishery patterns, demographic indices and spatial distribution of European hake, Merluccius -

Final EA Consist of a Corresponding Program at a Different Time with Issuance of an Associated IHA and the No Action Alternative, with No IHA and No Seismic Survey
Final Environmental Assessment of a Marine Geophysical Survey by the R/V Marcus G. Langseth in the Atlantic Ocean off Cape Hatteras, September–October 2014 Prepared for Lamont-Doherty Earth Observatory 61 Route 9W, P.O. Box 1000 Palisades, NY 10964-8000 and National Science Foundation Division of Ocean Sciences 4201 Wilson Blvd., Suite 725 Arlington, VA 22230 by LGL Ltd., environmental research associates 22 Fisher St., POB 280 King City, Ont. L7B 1A6 12 September 2014 LGL Report TA8350-1 Final Environmental Analysis for L-DEO Atlantic off Cape Hatteras, 2014 Page ii Table of Contents TABLE OF CONTENTS Page ABSTRACT ........................................................................................................................... V LIST OF ACRONYMS ......................................................................................................... VII I. PURPOSE AND NEED ....................................................................................................... 1 Mission of NSF ......................................................................................................................1 Purpose of and Need for the Proposed Action ...................................................................1 Background of NSF-funded Marine Seismic Research ....................................................1 Regulatory Setting ................................................................................................................2 II. ALTERNATIVES INCLUDING PROPOSED ACTION ....................................................... -
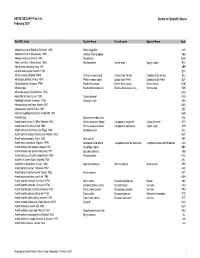
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

399 4. Bibliography
click for previous page 399 4. BIBLIOGRAPHY Alcock, A., 1889. Natural history notes from H.M. Indian Marine Survey Steamer “Investigator”, Commander Alfred Carpenter, R.N., D.S.O., commanding No. 13. On the bathybial fishes of the Bay of Bengal and neighboring waters, obtained during the seasons 1885-1889. Ann.Mag.Nat.Hist., ser. 6,6(23):376-399 .................., 1891. On the deep-sea fishes collected by the “Investigator” in 1890-1891. Ann.Mag.Nat.Hist., ser. 6,8:16-34; 119-138, pls vii-viii .................., 1899. A descriptive catalogue of the Indian deep-sea fishes in the Indian Museum. Being a revised account of the deep-sea fishes collected by the Royal Indian Marine Survey ship Investigator. Calcutta, Indian Museum, 211 pp. Allen, M.J. & G.8. Smith, 1988. Atlas and zoogeography of common fishes in the Bering Sea and Northeastern Pacific. NOAA Tech.Rep. NMFS, 66: 151 pp. Altukhov, K.A., 1979. O razmnozheznii i razvitii saiki Boreogadus saida (Lepechin) v Belom More. (The reproduction and development of the Arctic cod, Boreogadus saida, in the White Sea.) Vopr.lkhtiol.. 19(5):874-82 (J.Ichthyol., 19(5):93-101) Amaoka, K. et al. (eds), 1983. Fishes from the north-eastern Sea of Japan and the Okhotsk Sea off Hokkaido. Japan Fisheries Resource Conservation Association. Tokyo. 371 pp. Andriashev, A.P., 1954. Fishes of the northern seas of the USSR. Keys to the fauna of the USSR. Zool.lnst.USSR Acad.Sci., 53. Moscow-Leningrad, 617 p. (Transl. for Smithsonian Inst. and Nat.Sci.Found., by Israel Program for Sci.Transl., 1964) ...................., 1965. -

Fish, Crustaceans, Molluscs, Etc Capture Production by Species
488 Fish, crustaceans, molluscs, etc Capture production by species items Atlantic, Eastern Central C-34 Poissons, crustacés, mollusques, etc Captures par catégories d'espèces Atlantique, centre-est (a) Peces, crustáceos, moluscos, etc Capturas por categorías de especies Atlántico, centro-oriental English name Scientific name Species group Nom anglais Nom scientifique Groupe d'espèces 2010 2011 2012 2013 2014 2015 2016 Nombre inglés Nombre científico Grupo de especies t t t t t t t Tilapias nei Oreochromis (=Tilapia) spp 12 2 261 2 669 2 857 2 039 2 137 1 775 1 664 European eel Anguilla anguilla 22 9 ... 0 0 1 - - Shads nei Alosa spp 24 2 8 0 0 1 1 1 West African ilisha Ilisha africana 24 15 088 15 025 15 193 16 181 13 967 22 883 14 239 Mediterranean scaldfish Arnoglossus laterna 31 - - - - - - 57 Lefteye flounders nei Bothidae 31 46 ... 0 193 146 176 28 Common sole Solea solea 31 3 386 2 366 2 223 4 221 4 810 4 400 3 632 Sand sole Solea lascaris 31 ... ... 10 10 6 1 5 Wedge sole Dicologlossa cuneata 31 221 81 6 146 100 29 11 Soles nei Soleidae 31 5 264 6 167 8 273 9 313 10 575 6 830 8 290 Elongate tonguesole Symphurus ligulatus 31 - - - - - - 8 Tonguefishes Cynoglossidae 31 14 029 18 139 20 944 21 930 23 577 24 235 17 813 Megrim Lepidorhombus whiffiagonis 31 270 308 1 0 0 - 0 Turbot Psetta maxima 31 58 50 52 57 75 100 110 Turbots nei Scophthalmidae 31 ... ... ... ... ... ... 298 Citharids nei Citharidae 31 207 453 593 574 375 135 86 Spottail spiny turbot Psettodes belcheri 31 - 1 2 .. -

Merluccius Rafinesque, 1810 MERLU Merlu
click for previous page 327 Local Names : AUSTRALIA: Blue grenadier; GERMANY: Langschwanz-Seehecht; ITALY : Nasello azzurro; JAPAN : Hoki; NEW ZEALAND : Blue hake, Hoki, Whiptail; SPAIN : Merluza azul; USA : New Zealand whiptail. Literature : Armitage et al. (1981); Ayling & Cox (1982); Last et al. (1983). Remarks : Some specimens of Macruronus recently caught off western North Australia (18° S) might represent an undescribed species (N. Sinclair, pers. comm.). Merluccius Rafinesque, 1810 MERLU Merlu Genus with Reference : Merluccius Rafinesque, 1810:25. Type-species; Merluccius smiridus Rafinesque, 1810 (= Gadus merluccius Linnaeus, 1758) by monotypy. Diagnostic Features : Head large, about 1/3 to 1/4 of body length. Mouth large and oblique; maxillary reaching below middle of eye or behind it, almost half the length of head; lower jaw projecting below the upper; snout long and depressed, its length 1.3 to 3.2 times the eye diameter, its tip broad and rounded; eye large, its length 1/2 to 1/5 of upper jaw length; interorbital space broad, slightly elevated, its width 1.0 to 2.4 times the eye diameter; teeth in both jaws well developed, sharp, in two irregular rows; outer teeth fixed; inner ones larger and inwardly depressible; vomer with a biserial row of smaller teeth; no teeth on palatines; gill rakers well developed, varying in shape and number by species. Two separate dorsal fins, the first short, higher and triangular; the second long and partially divided by a notch at midlength; anal fin similar to second dorsal; pectoral fins long, slender and high in position, their relative length becoming smaller with growth; pelvic fins with 7 rays, placed in front of pectorals; caudal fin smaller than head and becoming progressively forked with growth; caudal skeleton possessing a set of X-Y bones.