Molecular-Pathogenetic Classification of Genetic Disorders of the Skeleton
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
Enzymes and Rna Complexes
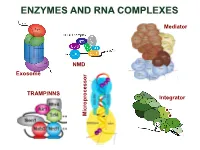
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

Genetics of Congenital Hand Anomalies
G. C. Schwabe1 S. Mundlos2 Genetics of Congenital Hand Anomalies Die Genetik angeborener Handfehlbildungen Original Article Abstract Zusammenfassung Congenital limb malformations exhibit a wide spectrum of phe- Angeborene Handfehlbildungen sind durch ein breites Spektrum notypic manifestations and may occur as an isolated malforma- an phänotypischen Manifestationen gekennzeichnet. Sie treten tion and as part of a syndrome. They are individually rare, but als isolierte Malformation oder als Teil verschiedener Syndrome due to their overall frequency and severity they are of clinical auf. Die einzelnen Formen kongenitaler Handfehlbildungen sind relevance. In recent years, increasing knowledge of the molecu- selten, besitzen aber aufgrund ihrer Häufigkeit insgesamt und lar basis of embryonic development has significantly enhanced der hohen Belastung für Betroffene erhebliche klinische Rele- our understanding of congenital limb malformations. In addi- vanz. Die fortschreitende Erkenntnis über die molekularen Me- tion, genetic studies have revealed the molecular basis of an in- chanismen der Embryonalentwicklung haben in den letzten Jah- creasing number of conditions with primary or secondary limb ren wesentlich dazu beigetragen, die genetischen Ursachen kon- involvement. The molecular findings have led to a regrouping of genitaler Malformationen besser zu verstehen. Der hohe Grad an malformations in genetic terms. However, the establishment of phänotypischer Variabilität kongenitaler Handfehlbildungen er- precise genotype-phenotype correlations for limb malforma- schwert jedoch eine Etablierung präziser Genotyp-Phänotyp- tions is difficult due to the high degree of phenotypic variability. Korrelationen. In diesem Übersichtsartikel präsentieren wir das We present an overview of congenital limb malformations based Spektrum kongenitaler Malformationen, basierend auf einer ent- 85 on an anatomic and genetic concept reflecting recent molecular wicklungsbiologischen, anatomischen und genetischen Klassifi- and developmental insights. -

A Disease-Linked Lncrna Mutation in Rnase MRP Inhibits Ribosome Synthesis
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A disease-linked lncRNA mutation in RNase MRP inhibits ribosome synthesis Nic Roberston1, Vadim Shchepachev1, David Wright2, Tomasz W. Turowski1, Christos Spanos1, Aleksandra Helwak1, Rose Zamoyska2, David Tollervey1 1 Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh, UK 2 Ashworth Laboratories, Institute of Immunology and Infection Research, University of Edinburgh, Edinburgh, UK Keywords: protein-RNA interaction; RNA-binding sites; UV crosslinking; mass spectrometry; genetic disease; Cartilage Hair Hypoplasia; ribosome synthesis; T cell activation Running title: RNase MRP defects cause ribosomopathy Highlights: • Mutations in RMRP lncRNA impair pre-rRNA processing and T cell activation • Patient derived fibroblasts show impaired pre-rRNA processing • Cells with the most common disease-linked mutation have specific processing defects • Cytoplasmic ribosomes and intact RNase MRP complexes are also reduced in these cells 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.29.437572; this version posted March 29, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Mutations in the human RMRP gene cause Cartilage Hair Hypoplasia (CHH), an autosomal recessive disorder characterized by skeletal abnormalities and impaired T cell activation. -

A Specialized Processing Body That Is Temporally and Asymmetrically Regulated During the Cell Cycle in Saccharomyces Cerevisiae
JCB: ARTICLE A specialized processing body that is temporally and asymmetrically regulated during the cell cycle in Saccharomyces cerevisiae Tina Gill, Jason Aulds, and Mark E. Schmitt Department of Biochemistry and Molecular Biology, State University of New York Upstate Medical University, Syracuse, NY 13210 Nase mitochondrial RNA processing (MRP) is an this spot was asymmetrically found in daughter cells, essential ribonucleoprotein endoribonuclease that where the RNase MRP substrate, CLB2 mRNA, localizes. R functions in the degradation of specifi c mRNAs in- Both the mitotic exit network and fourteen early ana- volved in cell cycle regulation. We have investigated phase release pathways are nonessential but important where this processing event occurs and how it is regu- for the temporal changes in localization. Asymmetric lo- lated. As expected, results demonstrate that RNase MRP calization was found to be dependent on the locasome. is predominantly localized in the nucleolus, where it The evidence suggests that these spots are specialized processes ribosomal RNAs. However, after the initiation processing bodies for the degradation of transcripts that of mitosis, RNase MRP localizes throughout the entire are cell cycle regulated and daughter cell localized. We nucleus and in a single discrete cytoplasmic spot that have called these TAM bodies for temporal asymmetric persists until the completion of telophase. Furthermore, MRP bodies. Introduction RNase mitochondrial RNA processing (MRP) is an essential ri- 27SA preribosomal RNA at the A3 site, forming the 5.8S(s) bonucleoprotein endoribonuclease that cleaves RNA substrates ribosomal RNA (rRNA; Schmitt and Clayton, 1993; Lygerou in a site-specifi c manner and is highly conserved in eukaryotes et al., 1996). -

The Rnase MRP and Rnase P Complexes, Will Be Summarised
PDF hosted at the Radboud Repository of the Radboud University Nijmegen The following full text is a publisher's version. For additional information about this publication click this link. http://hdl.handle.net/2066/19147 Please be advised that this information was generated on 2021-09-28 and may be subject to change. The human The human RNase MRP complex RNase MRP complex Composition, assembly and role in human disease Hans van Eenennaam Hans van Eenennaam The human RNase MRP complex Composition, assembly and role in human disease Hans van Eenennaam, 2002 The human RNase MRP complex Composition, assembly and role in human disease een wetenschappelijke proeve op het gebied van de Natuurwetenschappen, Wiskunde en Informatica PROEFSCHRIFT ter verkrijging van de graad van doctor aan de Katholieke Universiteit Nijmegen, volgens besluit van het College van Decanen in het openbaar te verdedigen op vrijdag 14 juni 2002 des namiddags om 1:30 uur precies door Hans van Eenennaam Cover illustration Statue of the Dwarf Seneb and his family, painted limestone, height 34 cm, width 22.5 cm, geboren op 3 november 1973 Giza, Tomb of Seneb, Late Fifth - Early Sixth te Middelburg Dynasty. From: The Cairo museum Masterpieces of Egyptian Art, Francesco Tiradritti, Thames & Hudson Ltd, London, 1998 Promotor Prof. Dr. W.J. van Venrooij Co-promotor Dr. G.J.M. Pruijn voor mijn ouders Manuscriptcommissie Prof. Dr. L.B.A. van de Putte Prof. Dr. F.P.J.T. Rutjes Prof. Dr. H.F. Tabak (Universiteit van Amsterdam) ISBN 90-9015696-8 © 2002 by Hans van Eenennaam The research described in this thesis was performed at the Department of Biochemistry, Faculty of Science, University of Nijmegen, the Netherlands. -

A Novel Model for the Rnase MRP-Induced Switch Between the Different Forms of 5.8S Rrna
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 29 April 2021 doi:10.20944/preprints202104.0765.v1 Research article A novel model for the RNase MRP-induced switch between the different forms of 5.8S rRNA Xiao Li1,2,3, Janice M Zengel1 and Lasse Lindahl1 1 Department of Biological Sciences, University of Maryland Baltimore County (UMBC) 1000 Hilltop Circle, Baltimore, Maryland 21250, USA 2 Department of Biology, University of Rochester, Rochester, New York 14627, USA 3 Current address: Abbott Laboratories, San Diego, CA ribosome biogenesis; rRNA processing; RNase MRP; long/short 5.8S rRNA © 2021 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 29 April 2021 doi:10.20944/preprints202104.0765.v1 Abstract: Processing of the RNA polymerase I pre-rRNA transcript into the mature 18S, 5.8S, and 25S rRNAs requires removing the “spacer” sequences. The canonical pathway for the removal of the ITS1 spacer, located between 18S and 5.8S rRNAs in the primary transcript, involves cleavages at the 3’ end of 18S rRNA and at two sites inside ITS1. The process generates a long and a short 5.8S rRNA that differ in the number of ITS1 nucleotides retained at the 5.8S 5’ end. Here we document a novel pathway that generates the long 5.8S for ITS1 while bypassing cleavage within ITS1. It entails a single endonuclease cut at the 3’-end of 18S rRNA followed by exonuclease Xrn1 degradation of ITS1. Mutations in RNase MRP increase the accumulation of long relative to short 5.8S rRNA; traditionally this is attributed to a decreased rate of RNase MRP cleavage at its target in ITS1, called A3. -

(12) Patent Application Publication (10) Pub. No.: US 2010/0210567 A1 Bevec (43) Pub
US 2010O2.10567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0210567 A1 Bevec (43) Pub. Date: Aug. 19, 2010 (54) USE OF ATUFTSINASATHERAPEUTIC Publication Classification AGENT (51) Int. Cl. A638/07 (2006.01) (76) Inventor: Dorian Bevec, Germering (DE) C07K 5/103 (2006.01) A6IP35/00 (2006.01) Correspondence Address: A6IPL/I6 (2006.01) WINSTEAD PC A6IP3L/20 (2006.01) i. 2O1 US (52) U.S. Cl. ........................................... 514/18: 530/330 9 (US) (57) ABSTRACT (21) Appl. No.: 12/677,311 The present invention is directed to the use of the peptide compound Thr-Lys-Pro-Arg-OH as a therapeutic agent for (22) PCT Filed: Sep. 9, 2008 the prophylaxis and/or treatment of cancer, autoimmune dis eases, fibrotic diseases, inflammatory diseases, neurodegen (86). PCT No.: PCT/EP2008/007470 erative diseases, infectious diseases, lung diseases, heart and vascular diseases and metabolic diseases. Moreover the S371 (c)(1), present invention relates to pharmaceutical compositions (2), (4) Date: Mar. 10, 2010 preferably inform of a lyophilisate or liquid buffersolution or artificial mother milk formulation or mother milk substitute (30) Foreign Application Priority Data containing the peptide Thr-Lys-Pro-Arg-OH optionally together with at least one pharmaceutically acceptable car Sep. 11, 2007 (EP) .................................. O7017754.8 rier, cryoprotectant, lyoprotectant, excipient and/or diluent. US 2010/0210567 A1 Aug. 19, 2010 USE OF ATUFTSNASATHERAPEUTIC ment of Hepatitis BVirus infection, diseases caused by Hepa AGENT titis B Virus infection, acute hepatitis, chronic hepatitis, full minant liver failure, liver cirrhosis, cancer associated with Hepatitis B Virus infection. 0001. The present invention is directed to the use of the Cancer, Tumors, Proliferative Diseases, Malignancies and peptide compound Thr-Lys-Pro-Arg-OH (Tuftsin) as a thera their Metastases peutic agent for the prophylaxis and/or treatment of cancer, 0008. -

Subject Index Proc
13088 Subject Index Proc. Natl. Acad. Sci. USA 91 (1994) Apoptosis in substantia nigra following developmental striatal excitotoxic Brine shrimp injury, 8117 See Artemia Visualizing hippocampal synaptic function by optical detection of Ca2l Broccol entry through the N-methyl-D-aspartate channel, 8170 See Brassica Amygdala modulation of hippocampal-dependent and caudate Bromophenacyl bromide nucleus-dependent memory processes, 8477 Bromophenacyl bromide binding to the actin-bundling protein I-plastin Distribution of corticotropin-releasing factor receptor mRNA expression inhibits inositol trisphosphate-independent increase in Ca2l in human in the rat brain and pituitary, 8777 neutrophils, 3534 Brownian dynamics Preproenkephalin promoter yields region-specific and long-term Adhesion of hard spheres under the influence of double-layer, van der expression in adult brain after direct in vivo gene transfer via a Waals, and gravitational potentials at a solid/liquid interface, 3004 defective herpes simplex viral vector, 8979 Browsers Intravenous administration of a transferrin receptor antibody-nerve Thorn-like prickles and heterophylly in Cyanea: Adaptations to extinct growth factor conjugate prevents the degeneration of cholinergic avian browsers on Hawaii?, 2810 striatal neurons in a model of Huntington disease, 9077 Bruton agammaglobulinemia Axotomy induces the expression of vasopressin receptors in cranial and Genomic organization and structure of Bruton agammaglobulinemia spinal motor nuclei in the adult rat, 9636 tyrosine kinase: Localization -

Human Ribonuclease P: Subunits, Function, and Intranuclear Localization
Downloaded from rnajournal.cshlp.org on September 23, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (2002), 8:1–7+ Cambridge University Press+ Printed in the USA+ Copyright © 2002 RNA Society+ DOI: 10+1017+S1355838201011189 REVIEW Human ribonuclease P: Subunits, function, and intranuclear localization NAYEF JARROUS Department of Molecular Biology, The Hebrew University–Hadassah Medical School, Jerusalem 91120, Israel ABSTRACT Catalytic complexes of nuclear ribonuclease P (RNase P) ribonucleoproteins are composed of several protein sub- units that appear to have specific roles in enzyme function in tRNA processing. This review describes recent progress made in the characterization of human RNase P, its relationship with the ribosomal RNA processing ribonucleoprotein RNase MRP, and the unexpected evolutionary conservation of its subunits. A new model for the biosynthesis of human RNase P is presented, in which this process is dynamic, transcription-dependent, and implicates functionally distinct nuclear compartments in tRNA biogenesis. Keywords: Cajal bodies; catalytic ribonucleoprotein; nucleolus; RNase MRP; RNase P; tRNA RIBONUCLEOPROTEIN COMPLEXES a ribonucleoprotein complex (Rossmanith et al+, 1995; OF RNase P Rossmanith & Karwan, 1998)+ These opposing find- ings provoke further debate on the biochemical nature Extensive purification analysis of RNase P from HeLa of mitochondrial and other organellar RNase P en- cells has revealed that the nuclear form of this tRNA zymes (Frank & Pace, 1998; Schon, 1999; Altman et al+, -

REVIEW ARTICLE Genetic Disorders of the Skeleton: a Developmental Approach
Am. J. Hum. Genet. 73:447–474, 2003 REVIEW ARTICLE Genetic Disorders of the Skeleton: A Developmental Approach Uwe Kornak and Stefan Mundlos Institute for Medical Genetics, Charite´ University Hospital, Campus Virchow, Berlin Although disorders of the skeleton are individually rare, they are of clinical relevance because of their overall frequency. Many attempts have been made in the past to identify disease groups in order to facilitate diagnosis and to draw conclusions about possible underlying pathomechanisms. Traditionally, skeletal disorders have been subdivided into dysostoses, defined as malformations of individual bones or groups of bones, and osteochondro- dysplasias, defined as developmental disorders of chondro-osseous tissue. In light of the recent advances in molecular genetics, however, many phenotypically similar skeletal diseases comprising the classical categories turned out not to be based on defects in common genes or physiological pathways. In this article, we present a classification based on a combination of molecular pathology and embryology, taking into account the importance of development for the understanding of bone diseases. Introduction grouping of conditions that have a common molecular origin but that have little in common clinically. For ex- Genetic disorders affecting the skeleton comprise a large ample, mutations in COL2A1 can result in such diverse group of clinically distinct and genetically heterogeneous conditions as lethal achondrogenesis type II and Stickler conditions. Clinical manifestations range from neonatal dysplasia, which is characterized by moderate growth lethality to only mild growth retardation. Although they retardation, arthropathy, and eye disease. It is now be- are individually rare, disorders of the skeleton are of coming increasingly clear that several distinct classifi- clinical relevance because of their overall frequency. -

Genome-Wide Investigation of Cellular Functions for Trna Nucleus
Genome-wide Investigation of Cellular Functions for tRNA Nucleus- Cytoplasm Trafficking in the Yeast Saccharomyces cerevisiae DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Hui-Yi Chu Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2012 Dissertation Committee: Anita K. Hopper, Advisor Stephen Osmani Kurt Fredrick Jane Jackman Copyright by Hui-Yi Chu 2012 Abstract In eukaryotic cells tRNAs are transcribed in the nucleus and exported to the cytoplasm for their essential role in protein synthesis. This export event was thought to be unidirectional. Surprisingly, several lines of evidence showed that mature cytoplasmic tRNAs shuttle between nucleus and cytoplasm and their distribution is nutrient-dependent. This newly discovered tRNA retrograde process is conserved from yeast to vertebrates. Although how exactly the tRNA nuclear-cytoplasmic trafficking is regulated is still under investigation, previous studies identified several transporters involved in tRNA subcellular dynamics. At least three members of the β-importin family function in tRNA nuclear-cytoplasmic intracellular movement: (1) Los1 functions in both the tRNA primary export and re-export processes; (2) Mtr10, directly or indirectly, is responsible for the constitutive retrograde import of cytoplasmic tRNA to the nucleus; (3) Msn5 functions solely in the re-export process. In this thesis I focus on the physiological role(s) of the tRNA nuclear retrograde pathway. One possibility is that nuclear accumulation of cytoplasmic tRNA serves to modulate translation of particular transcripts. To test this hypothesis, I compared expression profiles from non-translating mRNAs and polyribosome-bound translating mRNAs collected from msn5Δ and mtr10Δ mutants and wild-type cells, in fed or acute amino acid starvation conditions.