D-Phosphatidyl-Myo-Inositol 3-Phosphate
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-
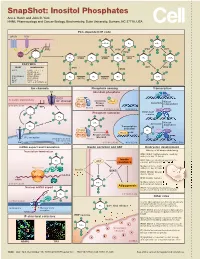
Snapshot: Inositol Phosphates Ace J
SnapShot: Inositol Phosphates Ace J. Hatch and John D. York HHMI, Pharmacology and Cancer Biology, Biochemistry, Duke University, Durham, NC 27710, USA PLC-dependent IP code GPCR RTK O O O O O O 5-PP-IP4 IP4 5-IP7 O O O O O O PIP2 O IP6K O IP6K O VIP1 O O O 2 O ITPK1 O 13 O PLC 2 O O O O O O O O 4 6 13 O 5 IP3 IPMK IP4 IPMK IP5 IPK1 IP6 1,5-IP8 4 6 O O 5 O O O O O O O O ENZYMES O O O O O O YEAST MAMMALIAN IP3K VIP1 IP6K IPMK PLC1 PLCβ, γ, δ, ε, ζ, η O - IP3KA, B, C - ITPK1 (IP56K) O O O O O O O O IPK2(ARG82) IPMK (IPK2) IP4 IP3 IP4 1-IP7 IPK1 IPK1 (IP5K) INPP5 ITPK1 KCS1 IP6K1, 2, 3 O O O O O O VIP1 VIP1, 2 (PPIP5K1, 2) O O Ion channels Phosphate sensing Transcription Cl- Abundant phosphate MCM1 ARG80 CIC3 P PLASMA MEMBRANE - Pho80 Cl channel Pho4 Kinase Kinase Assembly Pho85 independent CYTOPLASM activity 2 O PIP2 Pho81 13 CYTOPLASM NUCLEUS IPK2 ARG81 4 6 Phosphate starvation MCM1-ArgR O 5 O complex O O IP4 O O O O O O O 1-IP7 Kinase Activation dependent IP3 O O Transcription O O O activated Pho80 IP4 O X Pho4 O O Pho85 Kinase activity IP receptor blocked O 3 ENDOPLASMIC Pho81 RETICULUM Ca2+ CYTOPLASM NUCLEUS NUCLEUS mRNA export and translation Insulin secretion and AKT Embryonic development Translation termination Effects of IP kinase deficiency O IPMK (IPK2): Multiple defects, death by embryonic day 10 (mice) O O Insulin IPK1: Cillia are shortened and immotile IP6 AKT resistance causing patterning defects (zebrash) O O Multiple defects, death by Ribosome O embryonic day 8.5 (mice) GleI eRF1 Insulin GSK3β Dbp5 ITPK1 (IP56K): Neural tube -

ITPK1 Mediates the Lipid-Independent Synthesis of Inositol Phosphates Controlled by Metabolism
ITPK1 mediates the lipid-independent synthesis of inositol phosphates controlled by metabolism Yann Desfougèresa, Miranda S. C. Wilsona, Debabrata Lahaa, Gregory J. Millerb, and Adolfo Saiardia,1 aMedical Research Council Laboratory for Molecular Cell Biology, University College London, WC1E 6BT London, United Kingdom; and bDepartment of Chemistry, The Catholic University of America, Washington, DC 20064 Edited by Solomon H. Snyder, The Johns Hopkins University School of Medicine, Baltimore, MD, and approved October 25, 2019 (received for review July 3, 2019) Inositol phosphates (IPs) comprise a network of phosphorylated The potential pathways to IP6-7-8 synthesis have a profound molecules that play multiple signaling roles in eukaryotes. IPs impact on how we interpret their signaling roles. If they are synthesis is believed to originate with IP3 generated from PIP2 by synthesized from IP3, their function has evolved in relation to phospholipase C (PLC). Here, we report that in mammalian cells lipid-dependent PLC and/or calcium signaling. This constraint is PLC-generated IPs are rapidly recycled to inositol, and uncover the relieved if IP6 synthesis occurs lipid-independently, originating enzymology behind an alternative “soluble” route to synthesis of directly from inositol, a “soluble” pathway. In yeast, which do not IPs. Inositol tetrakisphosphate 1-kinase 1 (ITPK1)—found in Asgard have IP3-regulated calcium signaling, IP6 synthesis strictly depends archaea, social amoeba, plants, and animals—phosphorylates on the PLC-generated IP3 (Figs. 1A and 2D). The higher com- I(3)P1 originating from glucose-6-phosphate, and I(1)P1 generated plexity of other eukaryotes might allow different pathways for IPs from sphingolipids, to enable synthesis of IP6. -

Glycosylphosphatidylinositol Anchors from Galactomannan and GPI-Anchored Protein Are Synthesized by Distinct Pathways in Aspergillus Fumigatus
Journal of Fungi Article Glycosylphosphatidylinositol Anchors from Galactomannan and GPI-Anchored Protein Are Synthesized by Distinct Pathways in Aspergillus fumigatus Jizhou Li 1, Isabelle Mouyna 1, Christine Henry 1, Frédérique Moyrand 2, Christian Malosse 3, Julia Chamot-Rooke 3, Guilhem Janbon 2, Jean-Paul Latgé 1 and Thierry Fontaine 1,* 1 Unité des Aspergillus, 25 rue du Docteur Roux, Institut Pasteur, 25 rue du Docteur Roux, 75015 Paris, France; [email protected] (J.L.); [email protected] (I.M.); [email protected] (C.H.); [email protected] (J.-P.L.) 2 Unité de Biologie des ARN des Pathogènes Fongiques, Institut Pasteur, 25 rue du Docteur Roux, 75015 Paris, France; [email protected] (F.M.); [email protected] (G.J.) 3 Unité de Spectrométrie de Masse pour la Biologie, Institut Pasteur, CNRS USR 2000, 28 rue du Docteur Roux, 75015 Paris, France; [email protected] (C.M.); [email protected] (J.C.-R.) * Correspondence: [email protected]; Tel.: +33-145-688-358 Received: 8 December 2017; Accepted: 19 January 2018; Published: 2 Febuary 2018 Abstract: Glycosylphosphatidylinositols (GPIs) are lipid anchors allowing the exposure of proteins at the outer layer of the plasma membrane. In fungi, a number of GPI-anchored proteins (GPI-APs) are involved in the remodeling of the cell wall polymers. GPIs follow a specific biosynthetic pathway in the endoplasmic reticulum. After the transfer of the protein onto the GPI-anchor, a lipid remodeling occurs to substitute the diacylglycerol moiety by a ceramide. In addition to GPI-APs, A. fumigatus produces a GPI-anchored polysaccharide, the galactomannan (GM), that remains unique in the fungal kingdom. -

Dephosphorylation of 2,3-Bisphosphoglycerate by MIPP Expands the Regulatory Capacity of the Rapoport–Luebering Glycolytic Shunt
Dephosphorylation of 2,3-bisphosphoglycerate by MIPP expands the regulatory capacity of the Rapoport–Luebering glycolytic shunt Jaiesoon Cho*†, Jason S. King‡§, Xun Qian*, Adrian J. Harwood‡, and Stephen B. Shears*¶ *Laboratory of Signal Transduction, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Social Services, P.O. Box 12233, Research Triangle Park, NC 27709; and ‡Cardiff School of Biosciences, Cardiff University, Museum Avenue, Cardiff CF10 3US, United Kingdom Edited by Helen M. Ranney, University of California at San Diego, La Jolla, CA, and approved March 6, 2008 (received for review November 21, 2007) The Rapoport–Luebering glycolytic bypass comprises evolutionar- majority of mammals. By preferentially binding to deoxyhemo- ily conserved reactions that generate and dephosphorylate 2,3- globin, 2,3-BPG facilitates oxygen release from the erythrocyte bisphosphoglycerate (2,3-BPG). For >30 years, these reactions have to the surrounding tissues (6, 7). Thus, the regulation of eryth- been considered the responsibility of a single enzyme, the 2,3-BPG rocyte 2,3-BPG levels is key to efficiently meeting tissue oxygen synthase/2-phosphatase (BPGM). Here, we show that Dictyosteli- demands while also providing an important physiological adap- um, birds, and mammals contain an additional 2,3-BPG phospha- tation to oxygen deprivation (8), including that which occurs at tase that, unlike BPGM, removes the 3-phosphate. This discovery high altitude (9) or during postoperative anemia (10). reveals that the glycolytic pathway can bypass the formation of Despite the importance of carefully regulating 2,3-BPG turn- 3-phosphoglycerate, which is a precursor for serine biosynthesis over, little is known about how this might be achieved. -

Structural Basis for Phosphatidylinositol-Phosphate Biosynthesis
ARTICLE Received 8 Jun 2015 | Accepted 29 Aug 2015 | Published 16 Oct 2015 DOI: 10.1038/ncomms9505 OPEN Structural basis for phosphatidylinositol-phosphate biosynthesis Oliver B. Clarke1,*, David Tomasek2,*, Carla D. Jorge3,*, Meagan Belcher Dufrisne2, Minah Kim2, Surajit Banerjee4, Kanagalaghatta R. Rajashankar4, Lawrence Shapiro1, Wayne A. Hendrickson1, Helena Santos3 & Filippo Mancia2 Phosphatidylinositol is critical for intracellular signalling and anchoring of carbohydrates and proteins to outer cellular membranes. The defining step in phosphatidylinositol biosynthesis is catalysed by CDP-alcohol phosphotransferases, transmembrane enzymes that use CDP-diacylglycerol as donor substrate for this reaction, and either inositol in eukaryotes or inositol phosphate in prokaryotes as the acceptor alcohol. Here we report the structures of a related enzyme, the phosphatidylinositol-phosphate synthase from Renibacterium salmoninarum, with and without bound CDP-diacylglycerol to 3.6 and 2.5 Å resolution, respectively. These structures reveal the location of the acceptor site, and the molecular determinants of substrate specificity and catalysis. Functional characterization of the 40%-identical ortholog from Mycobacterium tuberculosis, a potential target for the development of novel anti-tuberculosis drugs, supports the proposed mechanism of substrate binding and catalysis. This work therefore provides a structural and functional framework to understand the mechanism of phosphatidylinositol-phosphate biosynthesis. 1 Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA. 2 Department of Physiology and Cellular Biophysics, Columbia University, New York, NY 10032, USA. 3 Biology Division, Instituto de Tecnologia Quı´mica e Biolo´gica, Universidade Nova de Lisboa, Avenida da Repu´blica-EAN, 2780-157 Oeiras, Portugal. 4 NE-CATand Department of Chemistry and Chemical Biology, Cornell University, Argonne National Laboratory, Argonne, IL 60439, USA. -

Glycosyl-Phosphatidylinositol/Inositol Phosphoglycan
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 8016-8019, September 1991 Biochemistry Glycosyl-phosphatidylinositol/inositol phosphoglycan: A signaling system for the low-affinity nerve growth factor receptor (development/inner ear/cochleovestibular ganglion/ant-ostol phosphoglycan antibody) JUAN REPRESA*, MATfAS A. AVILAt, CRISTINA MINERf, FERNANDO GIRALDEZt, GUILLERMO ROMERO§, ROSA CLEMENTEt, JOSE M. MATOt, AND ISABEL VARELA-NIETOt¶ *Departamento Ciencias Morfol6gicas and *Departamento Bioqutmica, Biologfa Molecular y Fisiologfa, Facultad de Medicina, Universidad de Valladolid, 47005 Valladolid, Spain; §Department of Pharmacology, University of Virginia, Charlottesville, VA 22908; and tInstituto de Investigaciones Biomddicas, Consejo Superior de Investigaciones Cientfficas and Departamento Bioqufmica, Universidad Aut6noma de Madrid, Arturo Duperier 4, 28029 Madrid, Spain Communicated by Sidney Udenfriend, June 7, 1991 (received for review April 15, 1991) ABSTRACT Nerve growth factor (NGF) exerts a variety of that IPG would be conserved for some of the developmental actions during embryonic development. At the early stages of actions of insulin and NGF, which could use a common inner ear development, NGF stimulates cell proliferation, an signaling pathway, shared perhaps with other related growth effect mediated through low-affinity receptors. We have stud- factors, to regulate cell growth. The present work provides ied the possibility that the glycosyl-phosphatidylinositol/ further support for the involvement of this glycosyl-PtdIns/ inositol phosphoglycan (glycosyl-Ptdlns/IPG) system is in- IPG pathway in transducing the mitogenic effects of NGF on volved in transmitting this NGF signal. Endogenous glycosyl- the early developing inner ear by showing the following PtdIns was characterized in extracts of cochleovestibular gan- results: (i) the presence of endogenous glycosyl-PtdIns and glia (CVGs) that incorporated [3Hglucosamine, [Hjglactose, IPG, the latter with strong mitogenic activity; (it) the ability [3lH]myristic acid, and PH]palmitic acid. -

Phosphatidylinositol 4,5-Bisphosphate Modifies Tubulin
The Journal of Neuroscience, March 1, 2002, 22(5):1668–1678 Phosphatidylinositol 4,5-Bisphosphate Modifies Tubulin  Participation in Phospholipase C 1 Signaling Juliana S. Popova,1 Arin K. Greene,1 Jia Wang,1 and Mark M. Rasenick1,2 Departments of 1Physiology and Biophysics and 2Psychiatry, University of Illinois at Chicago, College of Medicine, Chicago, Illinois 60612-7342 Tubulin forms the microtubule and regulates certain G-protein- the plasma membrane was demonstrated with confocal laser mediated signaling pathways. Both functions rely on the GTP- immunofluorescence microscopy. Although tubulin bound to ␣ ␣  binding properties of tubulin. Signal transduction through G q- both G q and PLC 1 , PIP2 facilitated the interaction between    ␣ regulated phospholipase C 1 (PLC 1 ) is activated by tubulin tubulin and PLC 1 but not that between tubulin and G q. ␣ ␣  through a direct transfer of GTP from tubulin to G q. However, However, PIP2 did augment formation of tubulin–G q–PLC 1   at high tubulin concentrations, inhibition of PLC 1 is observed. complexes. Subsequent to potentiating PLC 1 activation, sus-  This report demonstrates that tubulin inhibits PLC 1 by binding tained agonist-independent membrane binding of tubulin   ␣ the PLC 1 substrate phosphatidylinositol 4,5-bisphosphate at PIP2- and PLC 1-rich sites appeared to inhibit G q coupling  (PIP2 ). Tubulin binding of PIP2 was specific, because PIP2 but to PLC 1. Furthermore, colchicine increased membrane-  not phosphatidylinositol 3,4,5-trisphosphate, phosphatidylino- associated tubulin and also inhibited PLC 1 activity in SK- sitol 3-phosphate, phosphatidylinositol, phosphatidylcholine, N-SH cells. Thus, tubulin, depending on local membrane con- phosphatidylethanolamine, or inositol 1,4,5-trisphosphate in- centration, may serve as a positive or negative regulator of hibited microtubule assembly. -

IP3 Receptors – Lessons from Analyses Ex Cellula Ana M
© 2018. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs222463. doi:10.1242/jcs.222463 REVIEW SPECIAL ISSUE: RECONSTITUTING CELL BIOLOGY IP3 receptors – lessons from analyses ex cellula Ana M. Rossi and Colin W. Taylor* ABSTRACT and Ca2+ held within intracellular stores are entangled. For cardiac Inositol 1,4,5-trisphosphate receptors (IP Rs) are widely expressed muscle, depolarization of the plasma membrane (PM) causes 3 2+ intracellular channels that release Ca2+ from the endoplasmic voltage-gated Ca channels (Cav1.2, also known as CACNA1C) to 2+ reticulum (ER). We review how studies of IP Rs removed from their open, and the local increase in cytosolic free Ca concentration 3 2+ 2+ 2+ intracellular environment (‘ex cellula’), alongside similar analyses of ([Ca ]c) is then amplified by Ca -induced Ca release (CICR) ryanodine receptors, have contributed to understanding IP R through type 2 ryanodine receptors (RyR2) in the sarcoplasmic 3 2+ behaviour. Analyses of permeabilized cells have demonstrated that reticulum (Bers, 2002) (Fig. 1A). CICR and the local Ca 2+ signalling that is required to avoid CICR from becoming the ER is the major intracellular Ca store, and that IP3 stimulates Ca2+ release from this store. Radioligand binding confirmed that the explosive have become recurrent themes in the field of Ca2+ signalling (Rios, 2018). Fluorescent Ca2+ indicators and 4,5-phosphates of IP3 are essential for activating IP3Rs, and optical microscopy now allow Ca2+ sparks, local Ca2+ signals facilitated IP3R purification and cloning, which paved the way for evoked by a small cluster of RyRs, to be measured with exquisite structural analyses. -

Rapid Stimulation of Cellular Pi Uptake by the Inositol Pyrophosphate Insp8
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue Rapid stimulation of cellular Pi uptake by the inositol pyrophosphate InsP induced by its Cite this: Chem. Sci., 2020, 11,10265 8 All publication charges for this article photothermal release from lipid nanocarriers using have been paid for by the Royal Society † of Chemistry a near infra-red light-emitting diode Zhenzhen Wang,‡a Nikolaus Jork, b Tamara Bittner, b Huanchen Wang, a Henning J. Jessen b and Stephen B. Shears *a Inositol pyrophosphates (PP-InsPs), including diphospho-myo-inositol pentakisphosphate (5-InsP7) and bis-diphospho-myo-inositol tetrakisphosphate (1,5-InsP8), are highly polar, membrane-impermeant signaling molecules that control many homeostatic responses to metabolic and bioenergetic imbalance. To delineate their molecular activities, there is an increasing need for a toolbox of methodologies for real-time modulation of PP-InsP levels inside large populations of cultured cells. Here, we describe procedures to package PP-InsPs into thermosensitive phospholipid nanocapsules that are impregnated Creative Commons Attribution 3.0 Unported Licence. with a near infra-red photothermal dye; these liposomes are readily accumulated into cultured cells. The PP-InsPs remain trapped inside the liposomes until the cultures are illuminated with a near infra-red light-emitting diode (LED) which permeabilizes the liposomes to promote PP-InsP release. Additionally, so as to optimize these procedures, a novel stably fluorescent 5-InsP7 analogue (i.e., 5-FAM-InsP7) was synthesized with the assistance of click-chemistry; the delivery and deposition of the analogue inside cells was monitored by flow cytometry and by confocal microscopy. -

Role of Inositols and Inositol Phosphates in Energy Metabolism
molecules Review Role of Inositols and Inositol Phosphates in Energy Metabolism Saimai Chatree 1, Nanthaphop Thongmaen 2, Kwanchanit Tantivejkul 3, Chantacha Sitticharoon 2 and Ivana Vucenik 4,5,* 1 Faculty of Medicine and Public Health, HRH Princess Chulabhorn College of Medical Science, Chulabhorn Royal Academy, Bangkok 10210, Thailand; [email protected] 2 Department of Physiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand; [email protected] (N.T.); [email protected] (C.S.) 3 Sugavia Co., Ltd., Nakhonratchasima 30130, Thailand; [email protected] 4 Department of Medical and Research Technology, School of Medicine, University of Maryland, Baltimore, MD 21201, USA 5 Department of Pathology, School of Medicine, University of Maryland, Baltimore, MD 21201, USA * Correspondence: [email protected]; Tel.: +1-410-706-1832; Fax: +1-410-706-5229 Academic Editor: Stephen Shears Received: 3 October 2020; Accepted: 27 October 2020; Published: 1 November 2020 Abstract: Recently, inositols, especially myo-inositol and inositol hexakisphosphate, also known as phytic acid or IP6, with their biological activities received much attention for their role in multiple health beneficial effects. Although their roles in cancer treatment and prevention have been extensively reported, interestingly, they may also have distinctive properties in energy metabolism and metabolic disorders. We review inositols and inositol phosphate metabolism in mammalian cells to establish their biological activities and highlight their potential roles in energy metabolism. These molecules are known to decrease insulin resistance, increase insulin sensitivity, and have diverse properties with importance from cell signaling to metabolism. Evidence showed that inositol phosphates might enhance the browning of white adipocytes and directly improve insulin sensitivity through adipocytes. -

Lnositol 1,4,5=Trisphosphate Receptor Binding: Autoradiographic Localization in Rat Brain
The Journal of Neuroscience, January 1989, 9(l): 339-348 lnositol 1,4,5=Trisphosphate Receptor Binding: Autoradiographic Localization in Rat Brain Paul F. Worley,1s2 Jay M. Baraban,lv3 and Solomon H. Snyderi.3,4 Departments of ‘Neuroscience, *Neurology, 3Psychiatry and Behavioral Sciences, and 4Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205 lnositol 1,4,5trisphosphate is a second messenger gener- al., 1987a). In the present study, we have employed autora- ated by stimulation of the phosphoinositide cycle, thought diography to localize IP, receptors in brain in greater detail, to release calcium from intracellular stores. We have mapped comparing their distribution with that of PKC and adenylate the distribution of 3H-inositol 1,4,5trisphosphate receptor cyclase, labeled by 3H-phorbol 12,13-dibutyrate (3H-PDBu) and binding sites in rat brain by autoradiographic techniques. The 3H-forskolin, respectively. We have also characterized the on- cerebellum contains the highest level of inositol 1,4,5tris- togeny of IP, receptor binding in brain and performed lesion phosphate binding sites in brain, which appear to be selec- studies to identify neuronal elements enriched in IP, receptors. tively localized to Purkinje cells. Moderate levels of binding sites are present in the hippocampus, cerebral cortex, cau- Materials and Methods date, and substantia nigra. Lesion studies indicate that bind- *H-IP, autoradiography.3H-IP, autoradiography was carried out as de- ing in the hippocampus is restricted to intrinsic neuronal scribed (Worley et al., 1987a). Male Sprague-Dawley rats were anes- thetized with pentobarbital and perfused via the left cardiac ventricle elements and in the nigra is found on terminals of the stria- with buffer containing 50 mM sodium phosphate, pH 7.5, and 100 mM tonigral projection. -

Biochemical and Functional Responses Stimulated by Platelet-Activating Factor in Murine Peritoneal Macrophages
Biochemical and Functional Responses Stimulated by Platelet-activating Factor in Murine Peritoneal Macrophages Veronica Prpic,* Ronald J. Uhing,* James E. Weiel,* Lazlo Jakoi,~ Govind Gawdi,§ Brian Herman,§ and Dolph O. Adams*ll Departments of * Pathology, II Microbiology-Immunology, and eMedicine, Duke University, Durham, North Carolina 27710; and the §Department of Anatomy, University of North Carolina, Chapel Hill, North Carolina 27514 Abstract. Platelet-activating factor (PAF) is a potent the macrophages. PAF also led to increases of 1,2- stimulant of leukocytes, including macrophages. To diacylglycerol of • 200 pmol/107 cells. A characteristic analyze the mechanisms of its effects upon macro- pattern of enhanced protein phosphorylation, similar to Downloaded from http://rupress.org/jcb/article-pdf/107/1/363/1056534/363.pdf by guest on 02 February 2021 phages, we determined whether macrophages bear that initiated by both phorbol 12,13-myristate and lipo- specific surface receptors for PAE By competitive ra- polysaccharide, was observed and involved enhanced dioactive binding assays, we determined two classes of phosphorylation of proteins of 28, 33, 67, and 103 kD. specific receptors to be present on purified membranes The half-maximal dose of PAF for initiating all the derived from murine peritoneal macrophages (one hav- above effects was ,,,,5 x 10-9 M. PAF also initiated ing a Kd of '~1 X 10 -'° M and one a Ka of ~ 2 x 10-9 significant chemotaxis of the cells; the half-maximal M). When the macrophages were incubated with PAF, dose for this effect was ~1 x 10-" M. Taken together, rapid formation of several inositol phosphates includ- these observations suggest that murine mononuclear ing inositol 1,4,5-trisphosphate and inositol 1,3,4,5- phagocytes bear specific membrane receptors for PAF tetrakisphosphate were observed.