[5] Magnetic Densimetry: Partial Specific Volume and Other Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermodynamics Notes
Thermodynamics Notes Steven K. Krueger Department of Atmospheric Sciences, University of Utah August 2020 Contents 1 Introduction 1 1.1 What is thermodynamics? . .1 1.2 The atmosphere . .1 2 The Equation of State 1 2.1 State variables . .1 2.2 Charles' Law and absolute temperature . .2 2.3 Boyle's Law . .3 2.4 Equation of state of an ideal gas . .3 2.5 Mixtures of gases . .4 2.6 Ideal gas law: molecular viewpoint . .6 3 Conservation of Energy 8 3.1 Conservation of energy in mechanics . .8 3.2 Conservation of energy: A system of point masses . .8 3.3 Kinetic energy exchange in molecular collisions . .9 3.4 Working and Heating . .9 4 The Principles of Thermodynamics 11 4.1 Conservation of energy and the first law of thermodynamics . 11 4.1.1 Conservation of energy . 11 4.1.2 The first law of thermodynamics . 11 4.1.3 Work . 12 4.1.4 Energy transferred by heating . 13 4.2 Quantity of energy transferred by heating . 14 4.3 The first law of thermodynamics for an ideal gas . 15 4.4 Applications of the first law . 16 4.4.1 Isothermal process . 16 4.4.2 Isobaric process . 17 4.4.3 Isosteric process . 18 4.5 Adiabatic processes . 18 5 The Thermodynamics of Water Vapor and Moist Air 21 5.1 Thermal properties of water substance . 21 5.2 Equation of state of moist air . 21 5.3 Mixing ratio . 22 5.4 Moisture variables . 22 5.5 Changes of phase and latent heats . -
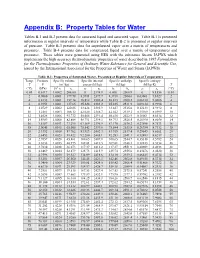
Appendix B: Property Tables for Water
Appendix B: Property Tables for Water Tables B-1 and B-2 present data for saturated liquid and saturated vapor. Table B-1 is presented information at regular intervals of temperature while Table B-2 is presented at regular intervals of pressure. Table B-3 presents data for superheated vapor over a matrix of temperatures and pressures. Table B-4 presents data for compressed liquid over a matrix of temperatures and pressures. These tables were generated using EES with the substance Steam_IAPWS which implements the high accuracy thermodynamic properties of water described in 1995 Formulation for the Thermodynamic Properties of Ordinary Water Substance for General and Scientific Use, issued by the International Associated for the Properties of Water and Steam (IAPWS). Table B-1: Properties of Saturated Water, Presented at Regular Intervals of Temperature Temp. Pressure Specific volume Specific internal Specific enthalpy Specific entropy T P (m3/kg) energy (kJ/kg) (kJ/kg) (kJ/kg-K) T 3 (°C) (kPa) 10 vf vg uf ug hf hg sf sg (°C) 0.01 0.6117 1.0002 206.00 0 2374.9 0.000 2500.9 0 9.1556 0.01 2 0.7060 1.0001 179.78 8.3911 2377.7 8.3918 2504.6 0.03061 9.1027 2 4 0.8135 1.0001 157.14 16.812 2380.4 16.813 2508.2 0.06110 9.0506 4 6 0.9353 1.0001 137.65 25.224 2383.2 25.225 2511.9 0.09134 8.9994 6 8 1.0729 1.0002 120.85 33.626 2385.9 33.627 2515.6 0.12133 8.9492 8 10 1.2281 1.0003 106.32 42.020 2388.7 42.022 2519.2 0.15109 8.8999 10 12 1.4028 1.0006 93.732 50.408 2391.4 50.410 2522.9 0.18061 8.8514 12 14 1.5989 1.0008 82.804 58.791 2394.1 58.793 2526.5 -

Equation of State of an Ideal Gas
ASME231 Atmospheric Thermodynamics NC A&T State U Department of Physics Dr. Yuh-Lang Lin http://mesolab.org [email protected] Lecture 3: Equation of State of an Ideal Gas As mentioned earlier, the state of a system is the condition of the system (or part of the system) at an instant of time measured by its properties. Those properties are called state variables, such as T, p, and V. The equation which relates T, p, and V is called equation of state. Since they are related by the equation of state, only two of them are independent. Thus, all the other thermodynamic properties will depend on the state defined by two independent variables by state functions. PVT system: The simplest thermodynamic system consists of a fixed mass of a fluid uninfluenced by chemical reactions or external fields. Such a system can be described by the pressure, volume and temperature, which are related by an equation of state f (p, V, T) = 0, (2.1) 1 where only two of them are independent. From physical experiments, the equation of state for an ideal gas, which is defined as a hypothetical gas whose molecules occupy negligible space and have no interactions, may be written as p = RT, (2.2) or p = RT, (2.3) where -3 : density (=m/V) in kg m , 3 -1 : specific volume (=V/m=1/) in m kg . R: specific gas constant (R=R*/M), -1 -1 -1 -1 [R=287 J kg K for dry air (Rd), 461 J kg K for water vapor for water vapor (Rv)]. -

Chapter 3 3.4-2 the Compressibility Factor Equation of State
Chapter 3 3.4-2 The Compressibility Factor Equation of State The dimensionless compressibility factor, Z, for a gaseous species is defined as the ratio pv Z = (3.4-1) RT If the gas behaves ideally Z = 1. The extent to which Z differs from 1 is a measure of the extent to which the gas is behaving nonideally. The compressibility can be determined from experimental data where Z is plotted versus a dimensionless reduced pressure pR and reduced temperature TR, defined as pR = p/pc and TR = T/Tc In these expressions, pc and Tc denote the critical pressure and temperature, respectively. A generalized compressibility chart of the form Z = f(pR, TR) is shown in Figure 3.4-1 for 10 different gases. The solid lines represent the best curves fitted to the data. Figure 3.4-1 Generalized compressibility chart for various gases10. It can be seen from Figure 3.4-1 that the value of Z tends to unity for all temperatures as pressure approach zero and Z also approaches unity for all pressure at very high temperature. If the p, v, and T data are available in table format or computer software then you should not use the generalized compressibility chart to evaluate p, v, and T since using Z is just another approximation to the real data. 10 Moran, M. J. and Shapiro H. N., Fundamentals of Engineering Thermodynamics, Wiley, 2008, pg. 112 3-19 Example 3.4-2 ---------------------------------------------------------------------------------- A closed, rigid tank filled with water vapor, initially at 20 MPa, 520oC, is cooled until its temperature reaches 400oC. -

Ideal Gasses Is Known As the Ideal Gas Law
ESCI 341 – Atmospheric Thermodynamics Lesson 4 –Ideal Gases References: An Introduction to Atmospheric Thermodynamics, Tsonis Introduction to Theoretical Meteorology, Hess Physical Chemistry (4th edition), Levine Thermodynamics and an Introduction to Thermostatistics, Callen IDEAL GASES An ideal gas is a gas with the following properties: There are no intermolecular forces, except during collisions. All collisions are elastic. The individual gas molecules have no volume (they behave like point masses). The equation of state for ideal gasses is known as the ideal gas law. The ideal gas law was discovered empirically, but can also be derived theoretically. The form we are most familiar with, pV nRT . Ideal Gas Law (1) R has a value of 8.3145 J-mol1-K1, and n is the number of moles (not molecules). A true ideal gas would be monatomic, meaning each molecule is comprised of a single atom. Real gasses in the atmosphere, such as O2 and N2, are diatomic, and some gasses such as CO2 and O3 are triatomic. Real atmospheric gasses have rotational and vibrational kinetic energy, in addition to translational kinetic energy. Even though the gasses that make up the atmosphere aren’t monatomic, they still closely obey the ideal gas law at the pressures and temperatures encountered in the atmosphere, so we can still use the ideal gas law. FORM OF IDEAL GAS LAW MOST USED BY METEOROLOGISTS In meteorology we use a modified form of the ideal gas law. We first divide (1) by volume to get n p RT . V we then multiply the RHS top and bottom by the molecular weight of the gas, M, to get Mn R p T . -

TABLE A-2 Properties of Saturated Water (Liquid–Vapor): Temperature Table Specific Volume Internal Energy Enthalpy Entropy # M3/Kg Kj/Kg Kj/Kg Kj/Kg K Sat
720 Tables in SI Units TABLE A-2 Properties of Saturated Water (Liquid–Vapor): Temperature Table Specific Volume Internal Energy Enthalpy Entropy # m3/kg kJ/kg kJ/kg kJ/kg K Sat. Sat. Sat. Sat. Sat. Sat. Sat. Sat. Temp. Press. Liquid Vapor Liquid Vapor Liquid Evap. Vapor Liquid Vapor Temp. Њ v ϫ 3 v Њ C bar f 10 g uf ug hf hfg hg sf sg C O 2 .01 0.00611 1.0002 206.136 0.00 2375.3 0.01 2501.3 2501.4 0.0000 9.1562 .01 H 4 0.00813 1.0001 157.232 16.77 2380.9 16.78 2491.9 2508.7 0.0610 9.0514 4 5 0.00872 1.0001 147.120 20.97 2382.3 20.98 2489.6 2510.6 0.0761 9.0257 5 6 0.00935 1.0001 137.734 25.19 2383.6 25.20 2487.2 2512.4 0.0912 9.0003 6 8 0.01072 1.0002 120.917 33.59 2386.4 33.60 2482.5 2516.1 0.1212 8.9501 8 10 0.01228 1.0004 106.379 42.00 2389.2 42.01 2477.7 2519.8 0.1510 8.9008 10 11 0.01312 1.0004 99.857 46.20 2390.5 46.20 2475.4 2521.6 0.1658 8.8765 11 12 0.01402 1.0005 93.784 50.41 2391.9 50.41 2473.0 2523.4 0.1806 8.8524 12 13 0.01497 1.0007 88.124 54.60 2393.3 54.60 2470.7 2525.3 0.1953 8.8285 13 14 0.01598 1.0008 82.848 58.79 2394.7 58.80 2468.3 2527.1 0.2099 8.8048 14 15 0.01705 1.0009 77.926 62.99 2396.1 62.99 2465.9 2528.9 0.2245 8.7814 15 16 0.01818 1.0011 73.333 67.18 2397.4 67.19 2463.6 2530.8 0.2390 8.7582 16 17 0.01938 1.0012 69.044 71.38 2398.8 71.38 2461.2 2532.6 0.2535 8.7351 17 18 0.02064 1.0014 65.038 75.57 2400.2 75.58 2458.8 2534.4 0.2679 8.7123 18 19 0.02198 1.0016 61.293 79.76 2401.6 79.77 2456.5 2536.2 0.2823 8.6897 19 20 0.02339 1.0018 57.791 83.95 2402.9 83.96 2454.1 2538.1 0.2966 8.6672 20 21 0.02487 1.0020 -

Definition of the Ideal Gas
ON THE DEFINITION OF THE IDEAL GAS. By Edgar Bucidngham. I . Nature and purpose of the definition.—The notion of the ideal gas is that of a gas having particularly simple physical properties to which the properties of the real gases may be considered as approximations; or of a standard to which the real gases may be referred, the properties of the ideal gas being simply defined and the properties of the real gases being then expressible as the prop- erties of the standard plus certain corrections which pertain to the individual gases. The smaller these corrections the more nearly the real gas approaches to being in the "ideal state." This con- ception grew naturally from the fact that the earlier experiments on gases showed that they did not differ much in their physical properties, so that it was possible to define an ideal standard in such a way that the corrections above referred to should in fact all be ''small" in terms of the unavoidable errors of experiment. Such a conception would hardly arise to-day, or if it did, would not be so simple as that which has come down to us from earlier times when the art of experimenting upon gases was less advanced. It is evident that a quantitative definition of the ideal standard gas needs to be more or less complete and precise according to the nature of the problem under immediate consideration. If changes of temperature and of internal energy play no part, all that is usually needed is a standard relation between pressure and volume. -

Glossary of Terms In
Glossary of Terms in Powder and Bulk Technology Prepared by Lyn Bates ISBN 978-0-946637-12-6 The British Materials Handling Board Foreward. Bulk solids play a vital role in human society, permeating almost all industrial activities and dominating many. Bulk technology embraces many disciplines, yet does not fall within the domain of a specific professional activity such as mechanical or chemical engineering. It has emerged comparatively recently as a coherent subject with tools for quantifying flow related properties and the behaviour of solids in handling and process plant. The lack of recognition of the subject as an established format with monumental industrial implications has impeded education in the subject. Minuscule coverage is offered within most university syllabuses. This situation is reinforced by the acceptance of empirical maturity in some industries and the paucity of quality textbooks available to address its enormous scope and range of application. Industrial performance therefore suffers. The British Materials Handling Board perceived the need for a Glossary of Terms in Particle Technology as an introductory tool for non-specialists, newcomers and students in this subject. Co-incidentally, a draft of a Glossary of Terms in Particulate Solids was in compilation. This concept originated as a project of the Working Part for the Mechanics of Particulate Solids, in support of a web site initiative of the European Federation of Chemical Engineers. The Working Party decided to confine the glossary on the EFCE web site to terms relating to bulk storage, flow of loose solids and relevant powder testing. Lyn Bates*, the UK industrial representative to the WPMPS leading this Glossary task force, decided to extend this work to cover broader aspects of particle and bulk technology and the BMHB arranged to publish this document as a contribution to the dissemination of information in this important field of industrial activity. -

Thermodynamics
THERMODYNAMICS PROPERTIES OF SINGLE-COMPONENT SYSTEMS For an ideal gas, Pv = RT or PV = mRT, and Nomenclature P1v1/T1 = P2v2/T2, where 1. Intensive properties are independent of mass. P = pressure, 2. Extensive properties are proportional to mass. v = speci c volume, 3. Speci c properties are lowercase (extensive/mass). m = mass of gas, R = gas constant, and State Functions (properties) 2 Absolute Pressure, P (lbf/in or Pa) T = absolute temperature. Absolute Temperature, T (°R or K) V = volume Volume, V (ft3 or m3) R is speci c to each gas but can be found from 3 3 Speci c Volume, vVm= (ft /lbm or m /kg) = R R ^h, where Internal Energy, U (Btu or kJ) mol. wt Speci c Internal Energy, R = the universal gas constant uUm= (usually in Btu/lbm or kJ/kg) = 1,545 ft-lbf/(lbmol-°R) = 8,314 J/(kmol⋅K). Enthalpy, H (Btu or KJ) For ideal gases, c – c = R Speci c Enthalpy, p v h = u + Pv = H/m (usually in Btu/lbm or kJ/kg) Also, for ideal gases: bb2h ll2u Entropy, S (Btu/°R or kJ/K) 2 ==002 P TTv Speci c Entropy, s = S/m [Btu/(lbm-°R) or kJ/(kg•K)] Gibbs Free Energy, g = h – Ts (usually in Btu/lbm or kJ/kg) For cold air standard, heat capacities are assumed to be constant at their room temperature values. In that case, the Helmholz Free Energy, following are true: a = u – Ts (usually in Btu/lbm or kJ/kg) Δu = c ΔT; Δh = c ΔT = bl2h v p Heat Capacity at Constant Pressure, cp 2 Δ T P s = cp ln (T2 /T1) – R ln (P2 /P1); and 2 = blu Δs = c ln (T /T ) + R ln (v /v ). -

C201-Notes-Chap1.Pdf
10/8/09 Concentration Units Concentration Units Molarity: defined as moles of solute per Molality: defined as moles of solute per liter of solution mass of solvent The volume used in the denominator is the total In this case, the mass is only the mass of volume of the solution, not just the volume of the the solvent, not the total mass of the solvent solution However, the volume of the solute is often negligible compared to the solvent volume Concentration Units Concentration Units Mole fraction: mole fraction is defined as Parts-per-million (ppm) and parts-per billion the number of moles solute divided by (ppb) the total number of moles of all species in For dilute aqueous solutions, we can make the solution the assumption that the density of the solution is 1.00 g/mL ppm and ppb express the mass of a specific solute relative to the mass of the solvent Mole fraction is a unitless number Concentration Units Concentration Units Parts-per-million (ppm) and parts-per billion Parts-per-million (ppm) and parts-per billion (ppb) (ppb) ppm = µg solute/mL soln When referring to gaseous mixtures, we = mg solute/L soln usually compare the number of solute particles in a specific volume (not the mass of solute particles) ppb = ng solute/mL soln ppmv = µmol solute/total mol in volume = µg solute/L soln ppbv = nmol solute/total mol in volume 1 10/8/09 Aqueous Solutions Molarity A solution is composed of two parts: the Determine concentration of a solution in solute and the solvent. -

P = PRT Or Pv = RT
Department of Earth & Climate Sciences Name_____________________ San Francisco State University In-Class Exercise/Lab 4 and Homework 4: Understanding the Gas Law 100 points (Due Monday 23 February 2015) The “simplified” or conceptual (explained in class) Ideal Gas Law (Equation of State) is: p = ρRT or (1a, b) pV = RT where p is pressure, ρ is density, R is a constant called the “gas constant” T is temperature. In the alternate version at the bottom, V is specific volume, which is the volume a unit amount of Gas, in this case 1 kiloGram, occupies. You can just think of this as “volume”. In reality, specific volume and density are inversely related. 1 V = ρ (2) As you can reason out, in the MKS system, the units of specific volume are m3 kg-1 and of density, kg m-3. Answer in complete sentences and on separate sheets. Part A. Concept ForminG 1. Examine equation 1(a). In a situation in which temperature remains constant, are pressure and density directly or inversely related? Explain? (Answer in complete sentences) (10 points) In a situation in which temperature remains constant, pressure is directly related to density. Since R and T are constant, to keep the equality, as density goes up, pressure must go up. 2. Examine equation 1(b). In situation in which volume remains constant, are pressure and temperature directly or inversely related? Explain. (10 points) In a situation in which volume (density) remains constant, pressure and temperature are directly related. Since R and V (or rho) are constant, as temperature goes up, pressure must go up. -

THERMODYNAMICS PROPERTIES of PURE SUBSTANCES Pure Substance a Substance That Has a Fixed Chemical Composition Throughout Is Called Pure Substance
Thermodynamics THERMODYNAMICS PROPERTIES OF PURE SUBSTANCES Pure Substance A substance that has a fixed chemical composition throughout is called pure substance. Water, helium carbon dioxide, nitrogen are examples. It does not have to be a single chemical element just as long as it is homogeneous throughout, like air. A mixture of phases of two or more substance is can still a pure substance if it is homogeneous, like ice and water (solid and liquid) or water and steam (liquid and gas). Vapor Vapor Liquid Liquid Water Air (Pure substance) (Not a pure substance because the composition of liquid air is different from the composition of vapor air) Phases of a Pure Substance There are three principle phases – solid, liquid and gas, but a substance can have several other phases within the principle phase. Examples include solid carbon (diamond and graphite) and iron (three solid phases). Nevertheless, thermodynamics deals with the primary phases only. In general: - Solids have strongest molecular bonds. - Solids are closely packed three dimensional crystals. - Their molecules do not move relative to each other - Intermediate molecular bond strength - Liquid molecular spacing is comparable to solids but their molecules can float about in groups. - There is molecular order within the groups - Weakest molecular bond strength. - Molecules in the gas phases are far apart, they have no ordered structure - The molecules move randomly and collide with each other. - Their molecules are at higher energy levels, they must release large amounts of energy to condense or freeze. THERMODYNAMICS 1 PROPERTIES OF PURE SUBSTANCES Thermodynamics Phase – Change Processes Of Pure Substances At this point, it is important to consider the liquid to solid phase change process.