2Vj0 Lichtarge Lab 2006
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -
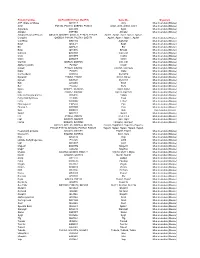
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures. -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -

Statistical Methods for Predicting Genetic Regulation by Nisar Ahmed
Statistical methods for predicting genetic regulation by Nisar Ahmed Shar Submitted in accordance with the requirements for the degree of Doctor of Philosophy The University of Leeds in the Faculty of Biological Sciences School of Molecular and Cellular Biology November 2016 ii Intellectual Property and Publication Statements The candidate confirms that the work submitted is his own, except where work which has formed part of jointly authored publications has been included. The contribution of the candidate and other authors to this work has been explicitly indicated below. The candidate confirms that appropriate credit has been given within the thesis where reference has been made to the work of others. The work in Chapter 5 and 6 of the thesis has appeared in publication as follows Shar, Nisar A., M. S. Vijayabaskar, and David R. Westhead. "Cancer somatic mutations cluster in a subset of regulatory sites predicted from the ENCODE data." Molecular Cancer 15.1 (2016): 76. I was responsible for carrying out this study and writing the paper along with the David R. Westhead. M.S. Vijayabaskar helped with data analysis, paper writing and supervision of the work. David R. Westhead conceived and designed the study. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. © 2016 The University of Leeds and Nisar Ahmed Shar The right of Nisar Ahmed Shar to be identified as Author of this work has been asserted by his in accordance with the copyright, Designs and Patents Act 1988. -

Integration of Protein Binding Interfaces and Abundance
INTEGRATION OF PROTEIN BINDING INTERFACES AND ABUNDANCE DATA REVEALS EVOLUTIONARY PRESSURES IN PROTEIN NETWORKS by David Orestis Holland A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy. Baltimore, Maryland January, 2018 © David O. Holland 2018 All rights reserved Abstract Networks of protein-protein interactions have received considerable interest in the past two decades for their insights about protein function and evolution. Traditionally, these networks only map the functional partners of proteins; they lack further levels of data such as binding affinity, allosteric regulation, competitive vs noncompetitive binding, and protein abundance. Recent experiments have made such data on a network-wide scale available, and in this thesis I integrate two extra layers of data in particular: the binding sites that proteins use to interact with their partners, and the abundance or “copy numbers” of the proteins. By analyzing the networks for the clathrin-mediated endocytosis (CME) system in yeast and the ErbB signaling pathway in humans, I find that this extra data reveals new insights about the evolution of protein networks. The structure of the binding site or interface interaction network (IIN) is optimized to allow higher binding specificity; that is, a high gap in strength between functional binding and nonfunctional mis-binding. This strongly implies that mis-binding is an evolutionary error-load constraint shaping protein network structure. Another method to limit mis-binding is to balance protein copy numbers so that there are no “leftover” proteins available for mis-binding. By developing a new method to quantify balance in IINs, I show that ii the CME network is significantly balanced when compared to randomly sampled sets of copy numbers. -

Gene Duplication, Genome Duplication, and the Functional Diversification of Vertebrate Globins Jay F
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Jay F. Storz Publications Papers in the Biological Sciences 2-2016 Gene duplication, genome duplication, and the functional diversification of vertebrate globins Jay F. Storz University of Nebraska - Lincoln, [email protected] Juan C. Opazo Universidad Austral de Chile, [email protected] Federico G. Hoffmann Mississippi State University, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/bioscistorz Storz, Jay F.; Opazo, Juan C.; and Hoffmann, Federico G., "Gene duplication, genome duplication, and the functional diversification of vertebrate globins" (2016). Jay F. Storz Publications. 70. http://digitalcommons.unl.edu/bioscistorz/70 This Article is brought to you for free and open access by the Papers in the Biological Sciences at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Jay F. Storz Publications by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. NIH Public Access Author Manuscript Mol Phylogenet Evol. Author manuscript; available in PMC 2015 January 26. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Phylogenet Evol. 2013 February ; 66(2): 469–478. doi:10.1016/j.ympev.2012.07.013. Gene duplication, genome duplication, and the functional diversification of vertebrate globins Jay F. Storza,*, Juan C. Opazob, and Federico G. Hoffmannc,d aSchool of Biological Sciences, University -

Tepzz 8Z6z54a T
(19) TZZ ZZ_T (11) EP 2 806 054 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 26.11.2014 Bulletin 2014/48 C40B 40/06 (2006.01) C12Q 1/68 (2006.01) C40B 30/04 (2006.01) C07H 21/00 (2006.01) (21) Application number: 14175049.7 (22) Date of filing: 28.05.2009 (84) Designated Contracting States: (74) Representative: Irvine, Jonquil Claire AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HGF Limited HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 140 London Wall PT RO SE SI SK TR London EC2Y 5DN (GB) (30) Priority: 28.05.2008 US 56827 P Remarks: •Thecomplete document including Reference Tables (62) Document number(s) of the earlier application(s) in and the Sequence Listing can be downloaded from accordance with Art. 76 EPC: the EPO website 09753364.0 / 2 291 553 •This application was filed on 30-06-2014 as a divisional application to the application mentioned (71) Applicant: Genomedx Biosciences Inc. under INID code 62. Vancouver, British Columbia V6J 1J8 (CA) •Claims filed after the date of filing of the application/ after the date of receipt of the divisional application (72) Inventor: Davicioni, Elai R.68(4) EPC). Vancouver British Columbia V6J 1J8 (CA) (54) Systems and methods for expression- based discrimination of distinct clinical disease states in prostate cancer (57) A system for expression-based discrimination of distinct clinical disease states in prostate cancer is provided that is based on the identification of sets of gene transcripts, which are characterized in that changes in expression of each gene transcript within a set of gene transcripts can be correlated with recurrent or non- recur- rent prostate cancer. -

Prevalence of Chromosomal Rearrangements Involving Non-ETS Genes in Prostate Cancer
INTERNATIONAL JOURNAL OF ONCOLOGY 46: 1637-1642, 2015 Prevalence of chromosomal rearrangements involving non-ETS genes in prostate cancer Martina KLUTH1*, RAMI GALAL1*, ANTJE KROHN1, JOACHIM WEISCHENFELDT4, CHRISTINA TSOURLAKIS1, LISA PAUSTIAN1, RAMIN Ahrary1, MALIK AHMED1, SEKANDER SCHERZAI1, ANNE MEYER1, HÜSEYIN SIRMA1, JAN KORBEL4, GUIDO SAUTER1, THORSTEN SCHLOMM2,3, RONALD SIMON1 and SARAH MINNER1 1Institute of Pathology, 2Martini-Clinic, Prostate Cancer Center, and 3Department of Urology, Section for Translational Prostate Cancer Research, University Medical Center Hamburg-Eppendorf; 4Genome Biology Unit, European Molecular Biology Laboratory (EMBL), D-69117 Heidelberg, Germany Received November 25, 2014; Accepted December 30, 2014 DOI: 10.3892/ijo.2015.2855 Abstract. Prostate cancer is characterized by structural rear- tumors that can be surgically treated in a curative manner, rangements, most frequently including translocations between ~20% of the tumors will progress to metastatic and hormone androgen-dependent genes and members of the ETS family refractory disease, accounting for >250.000 deaths per year of transcription factor like TMPRSS2:ERG. In a recent whole worldwide (1). Targeted therapies that would allow for an genome sequencing study we identified 140 gene fusions that effective treatment after failure of androgen withdrawal were unrelated to ETS genes in 11 prostate cancers. The aim therapy are lacking. of the present study was to estimate the prevalence of non-ETS Recent whole genome sequencing studies have shown that gene fusions. We randomly selected 27 of these rearrange- the genomic landscape of prostate cancer differs markedly ments and analyzed them by fluorescencein situ hybridization from that of other solid tumor types. Whereas, for example, (FISH) in a tissue microarray format containing 500 prostate breast or colon cancer is characterized by high-grade genetic cancers. -

PDF Output of CLIC (Clustering by Inferred Co-Expression)
PDF Output of CLIC (clustering by inferred co-expression) Dataset: Num of genes in input gene set: 6 Total number of genes: 16493 CLIC PDF output has three sections: 1) Overview of Co-Expression Modules (CEMs) Heatmap shows pairwise correlations between all genes in the input query gene set. Red lines shows the partition of input genes into CEMs, ordered by CEM strength. Each row shows one gene, and the brightness of squares indicates its correlations with other genes. Gene symbols are shown at left side and on the top of the heatmap. 2) Details of each CEM and its expansion CEM+ Top panel shows the posterior selection probability (dataset weights) for top GEO series datasets. Bottom panel shows the CEM genes (blue rows) as well as expanded CEM+ genes (green rows). Each column is one GEO series dataset, sorted by their posterior probability of being selected. The brightness of squares indicates the gene's correlations with CEM genes in the corresponding dataset. CEM+ includes genes that co-express with CEM genes in high-weight datasets, measured by LLR score. 3) Details of each GEO series dataset and its expression profile: Top panel shows the detailed information (e.g. title, summary) for the GEO series dataset. Bottom panel shows the background distribution and the expression profile for CEM genes in this dataset. Overview of Co-Expression Modules (CEMs) with Dataset Weighting Scale of average Pearson correlations Num of Genes in Query Geneset: 6. Num of CEMs: 1. 0.0 0.2 0.4 0.6 0.8 1.0 Epn1 Ap2a1 Ralbp1 Ap2a2 Numb Ccnb1 Epn1 Ap2a1 Ralbp1