Integration of Protein Binding Interfaces and Abundance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Regulation of Cdc42 and Its Effectors in Epithelial Morphogenesis Franck Pichaud1,2,*, Rhian F
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs217869. doi:10.1242/jcs.217869 REVIEW SUBJECT COLLECTION: ADHESION Regulation of Cdc42 and its effectors in epithelial morphogenesis Franck Pichaud1,2,*, Rhian F. Walther1 and Francisca Nunes de Almeida1 ABSTRACT An overview of Cdc42 Cdc42 – a member of the small Rho GTPase family – regulates cell Cdc42 was discovered in yeast and belongs to a large family of small – polarity across organisms from yeast to humans. It is an essential (20 30 kDa) GTP-binding proteins (Adams et al., 1990; Johnson regulator of polarized morphogenesis in epithelial cells, through and Pringle, 1990). It is part of the Ras-homologous Rho subfamily coordination of apical membrane morphogenesis, lumen formation and of GTPases, of which there are 20 members in humans, including junction maturation. In parallel, work in yeast and Caenorhabditis elegans the RhoA and Rac GTPases, (Hall, 2012). Rho, Rac and Cdc42 has provided important clues as to how this molecular switch can homologues are found in all eukaryotes, except for plants, which do generate and regulate polarity through localized activation or inhibition, not have a clear homologue for Cdc42. Together, the function of and cytoskeleton regulation. Recent studies have revealed how Rho GTPases influences most, if not all, cellular processes. important and complex these regulations can be during epithelial In the early 1990s, seminal work from Alan Hall and his morphogenesis. This complexity is mirrored by the fact that Cdc42 can collaborators identified Rho, Rac and Cdc42 as main regulators of exert its function through many effector proteins. -

Mouse Wipf1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Wipf1 Knockout Project (CRISPR/Cas9) Objective: To create a Wipf1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Wipf1 gene (NCBI Reference Sequence: NM_153138 ; Ensembl: ENSMUSG00000075284 ) is located on Mouse chromosome 2. 8 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 8 (Transcript: ENSMUST00000094681). Exon 3~6 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Homozygous mutants have immunological abnormalities, although lymphocyte development appears normal. Mutants show abnormal B and T cell proliferative responses, high serum immunoglobulin levels and impaired immunological synapse formation. Exon 3 starts from about 3.52% of the coding region. Exon 3~6 covers 85.26% of the coding region. The size of effective KO region: ~9629 bp. The KO region does not have any other known gene. Page 1 of 9 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 3 4 5 6 8 Legends Exon of mouse Wipf1 Knockout region Page 2 of 9 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 3 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -
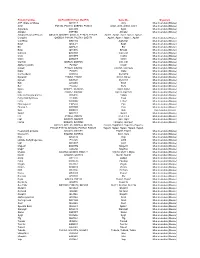
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -

The Guanine Nucleotide Exchange Factor Arhgef5 Plays Crucial Roles in Src-Induced Podosome Formation
1726 Research Article The guanine nucleotide exchange factor Arhgef5 plays crucial roles in Src-induced podosome formation Miho Kuroiwa, Chitose Oneyama, Shigeyuki Nada and Masato Okada* Department of Oncogene Research, Research institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan *Author for correspondence ([email protected]) Accepted 19 January 2011 Journal of Cell Science 124, 1726-1738 © 2011. Published by The Company of Biologists Ltd doi:10.1242/jcs.080291 Summary Podosomes and invadopodia are actin-rich membrane protrusions that play a crucial role in cell adhesion and migration, and extracellular matrix remodeling in normal and cancer cells. The formation of podosomes and invadopodia is promoted by upregulation of some oncogenic molecules and is closely related to the invasive potential of cancer cells. However, the molecular mechanisms underlying the podosome and invadopodium formation still remain unclear. Here, we show that a guanine nucleotide exchange factor (GEF) for Rho family GTPases (Arhgef5) is crucial for Src-induced podosome formation. Using an inducible system for Src activation, we found that Src-induced podosome formation depends upon the Src SH3 domain, and identified Arhgef5 as a Src SH3-binding protein. RNA interference (RNAi)-mediated depletion of Arhgef5 caused robust inhibition of Src-dependent podosome formation. Overexpression of Arhgef5 promoted actin stress fiber remodeling through activating RhoA, and the activation of RhoA or Cdc42 was required for Src-induced podosome formation. Arhgef5 was tyrosine-phosphorylated by Src and bound to Src to positively regulate its activity. Furthermore, the pleckstrin homology (PH) domain of Arhgef5 was required for podosome formation, and Arhgef5 formed a ternary complex with Src and phosphoinositide 3-kinase when Src and/or Arhgef5 were upregulated. -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures. -

Microrna Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton
cells Review MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton , , Karen Uray * y , Evelin Major and Beata Lontay * y Department of Medical Chemistry, Faculty of Medicine, University of Debrecen, 4032 Debrecen, Hungary; [email protected] * Correspondence: [email protected] (K.U.); [email protected] (B.L.); Tel.: +36-52-412345 (K.U. & B.L.) The authors contributed equally to the manuscript. y Received: 11 June 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: MicroRNAs (miRNAs) are key modulators of post-transcriptional gene regulation in a plethora of processes, including actin–myosin cytoskeleton dynamics. Recent evidence points to the widespread effects of miRNAs on actin–myosin cytoskeleton dynamics, either directly on the expression of actin and myosin genes or indirectly on the diverse signaling cascades modulating cytoskeletal arrangement. Furthermore, studies from various human models indicate that miRNAs contribute to the development of various human disorders. The potentially huge impact of miRNA-based mechanisms on cytoskeletal elements is just starting to be recognized. In this review, we summarize recent knowledge about the importance of microRNA modulation of the actin–myosin cytoskeleton affecting physiological processes, including cardiovascular function, hematopoiesis, podocyte physiology, and osteogenesis. Keywords: miRNA; actin; myosin; actin–myosin complex; Rho kinase; cancer; smooth muscle; hematopoiesis; stress fiber; gene expression; cardiovascular system; striated muscle; muscle cell differentiation; therapy 1. Introduction Actin–myosin interactions are the primary source of force generation in mammalian cells. Actin forms a cytoskeletal network and the myosin motor proteins pull actin filaments to produce contractile force. All eukaryotic cells contain an actin–myosin network inferring contractile properties to these cells. -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g.