Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

UTF1 Antibody A
Revision 1 C 0 2 - t UTF1 Antibody a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 9 0 Web: [email protected] 9 www.cellsignal.com 3 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source: UniProt ID: Entrez-Gene Id: WB M R Endogenous 40 Rabbit Q5T230 8433 Product Usage Information Application Dilution Western Blotting 1:1000 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA and 50% glycerol. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity UTF1 Antibody detects endogenous levels of total UTF1 protein. Species Reactivity: Mouse, Rat Species predicted to react based on 100% sequence homology: Human Source / Purification Polyclonal antibodies are produced by immunizing animals with a synthetic peptide correponding to amino acid sequence near the C-terminus of human UTF1. Antibodies are purified by Protein A and peptide affinity chromatography. Background Undifferentiated embryonic cell transcription factor 1 (UTF1) is expressed in cells of the inner cell mass and the epiblast (1). Expression is down-regulated with development, although it is maintained in the embryonic germ cells and in the adult gonads (1). Reduced expression in embryonic stem cells (ESCs) is associated with failure to differentiate properly, although self-renewal is unaffected (2). UTF1 is tightly associated with chromatin in mouse and human ESCs and may be involved in maintaining an epigenetic environment necessary for the pluripotent state (2,3). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives
cancers Review Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives Shirin Hafezi 1 and Mohamed Rahmani 1,2,* 1 Research Institute of Medical & Health Sciences, University of Sharjah, P.O. Box 27272 Sharjah, United Arab Emirates; [email protected] 2 Department of Basic Medical Sciences, College of Medicine, University of Sharjah, P.O. Box 27272 Sharjah, United Arab Emirates * Correspondence: [email protected]; Tel.: +971-6505-7759 Simple Summary: Apoptosis, a programmed form of cell death, represents the main mechanism by which cells die. Such phenomenon is highly regulated by the BCL-2 family of proteins, which includes both pro-apoptotic and pro-survival proteins. The decision whether cells live or die is tightly controlled by a balance between these two classes of proteins. Notably, the pro-survival Bcl-2 proteins are frequently overexpressed in cancer cells dysregulating this balance in favor of survival and also rendering cells more resistant to therapeutic interventions. In this review, we outlined the most important steps in the development of targeting the BCL-2 survival proteins, which laid the ground for the discovery and the development of the selective BCL-2 inhibitor venetoclax as a therapeutic drug in hematological malignancies. The limitations and future directions are also discussed. Abstract: The major form of cell death in normal as well as malignant cells is apoptosis, which is a programmed process highly regulated by the BCL-2 family of proteins. This includes the antiapoptotic proteins (BCL-2, BCL-XL, MCL-1, BCLW, and BFL-1) and the proapoptotic proteins, which can be divided into two groups: the effectors (BAX, BAK, and BOK) and the BH3-only proteins Citation: Hafezi, S.; Rahmani, M. -

H4K16 Acetylation Marks Active Genes and Enhancers of Embryonic Stem Cells, but Does Not Alter Chromatin Compaction
Downloaded from genome.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction Gillian Taylor1, Ragnhild Eskeland2, Betül Hekimoglu-Balkan1, Madapura M. Pradeepa1* and Wendy A Bickmore1* 1 MRC Human Genetics Unit, MRC Institute of Genetics and Molecular Medicine at University of Edinburgh, Crewe Road, Edinburgh EH4 2XU, UK 2Current address: Department of Molecular Biosciences, University of Oslo, N-0316 Oslo, Norway *Correspondence to: W. Bickmore or M.M. Pradeepa, MRC Human Genetics Unit, MRC IGMM, Crewe Road, Edinburgh EH4 2XU, UK Tel: +44 131 332 2471 Fax: +44 131 467 8456 Email:[email protected] or [email protected] Running head: H4K16 acetylation and long-range genome regulation Keywords: Chromatin compaction, embryonic stem cells, fluorescence in situ hybridization, histone acetylation, long-range regulation, 1 Downloaded from genome.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract Compared with histone H3, acetylation of H4 tails has not been well studied, especially in mammalian cells. Yet, H4K16 acetylation is of particular interest because of its ability to decompact nucleosomes in vitro and its involvement in dosage compensation in flies. Here we show that, surprisingly, loss of H4K16 acetylation does not alter higher-order chromatin compaction in vivo in mouse embryonic stem cells (ESCs). As well as peaks of acetylated H4K16 and Kat8/MOF histone acetyltransferase at the transcription start sites of expressed genes, we report that acetylation of H4K16 is a new marker of active enhancers in ESCs and that some enhancers are marked by H3K4me1, Kat8 and H4K16ac but not by acetylated H3K27 or p300/EP300, suggesting that they are novel EP300 independent regulatory elements. -
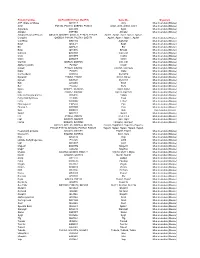
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -
![PADI4 Mouse Monoclonal Antibody [Clone ID: OTI5C10] Product Data](https://docslib.b-cdn.net/cover/0337/padi4-mouse-monoclonal-antibody-clone-id-oti5c10-product-data-660337.webp)
PADI4 Mouse Monoclonal Antibody [Clone ID: OTI5C10] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for CF504896 PADI4 Mouse Monoclonal Antibody [Clone ID: OTI5C10] Product data: Product Type: Primary Antibodies Clone Name: OTI5C10 Applications: IHC, WB Recommended Dilution: WB 1:2000, IHC 1:150 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment correspongding to amino acids 299-588 of human PADI4(NP_036519) produced in E.coli. Formulation: Lyophilized powder (original buffer 1X PBS, pH 7.3, 8% trehalose) Reconstitution Method: For reconstitution, we recommend adding 100uL distilled water to a final antibody concentration of about 1 mg/mL. To use this carrier-free antibody for conjugation experiment, we strongly recommend performing another round of desalting process. (OriGene recommends Zeba Spin Desalting Columns, 7KMWCO from Thermo Scientific) Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 73.9 kDa Gene Name: Homo sapiens peptidyl arginine deiminase 4 (PADI4), mRNA. Database Link: NP_036519 Entrez Gene 23569 Human Q9UM07 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 4 PADI4 Mouse Monoclonal Antibody [Clone ID: OTI5C10] – CF504896 Background: This gene is a member of a gene family which encodes enzymes responsible for the conversion of arginine residues to citrulline residues. -

Deimination, Intermediate Filaments and Associated Proteins
International Journal of Molecular Sciences Review Deimination, Intermediate Filaments and Associated Proteins Julie Briot, Michel Simon and Marie-Claire Méchin * UDEAR, Institut National de la Santé Et de la Recherche Médicale, Université Toulouse III Paul Sabatier, Université Fédérale de Toulouse Midi-Pyrénées, U1056, 31059 Toulouse, France; [email protected] (J.B.); [email protected] (M.S.) * Correspondence: [email protected]; Tel.: +33-5-6115-8425 Received: 27 October 2020; Accepted: 16 November 2020; Published: 19 November 2020 Abstract: Deimination (or citrullination) is a post-translational modification catalyzed by a calcium-dependent enzyme family of five peptidylarginine deiminases (PADs). Deimination is involved in physiological processes (cell differentiation, embryogenesis, innate and adaptive immunity, etc.) and in autoimmune diseases (rheumatoid arthritis, multiple sclerosis and lupus), cancers and neurodegenerative diseases. Intermediate filaments (IF) and associated proteins (IFAP) are major substrates of PADs. Here, we focus on the effects of deimination on the polymerization and solubility properties of IF proteins and on the proteolysis and cross-linking of IFAP, to finally expose some features of interest and some limitations of citrullinomes. Keywords: citrullination; post-translational modification; cytoskeleton; keratin; filaggrin; peptidylarginine deiminase 1. Introduction Intermediate filaments (IF) constitute a unique macromolecular structure with a diameter (10 nm) intermediate between those of actin microfilaments (6 nm) and microtubules (25 nm). In humans, IF are found in all cell types and organize themselves into a complex network. They play an important role in the morphology of a cell (including the nucleus), are essential to its plasticity, its mobility, its adhesion and thus to its function. -

Dynamics of the BH3-Only Protein Binding Interface of Bcl-Xl
Biophysical Journal Volume 109 September 2015 1049–1057 1049 Article Dynamics of the BH3-Only Protein Binding Interface of Bcl-xL Xiaorong Liu,1 Alex Beugelsdijk,1 and Jianhan Chen1,* 1Department of Biochemistry and Molecular Biophysics, Kansas State University, Manhattan, Kansas ABSTRACT The balance and interplay between pro-death and pro-survival members of the B-cell lymphoma-2 (Bcl-2) family proteins play key roles in regulation of the mitochondrial pathway of programmed cell death. Recent NMR and biochemical studies have revealed that binding of the proapoptotic BH3-only protein PUMA induces significant unfolding of antiapoptotic Bcl-xL at the interface, which in turn disrupts the Bcl-xL/p53 interaction to activate apoptosis. However, the molecular mecha- nism of such regulated unfolding of Bcl-xL is not fully understood. Analysis of the existing Protein Data Bank structures of Bcl-xL in both bound and unbound states reveal substantial intrinsic heterogeneity at its BH3-only protein binding interface. Large-scale atomistic simulations were performed in explicit solvent for six representative structures to further investigate the intrinsic confor- mational dynamics of Bcl-xL. The results support that the BH3-only protein binding interface of Bcl-xL is much more dynamic compared to the rest of the protein, both unbound and when bound to various BH3-only proteins. Such intrinsic interfacial confor- mational dynamics likely provides a physical basis that allows Bcl-xL to respond sensitively to detailed biophysical properties of the ligand. The ability of Bcl-xL to retain or even enhance dynamics at the interface in bound states could further facilitate the regulation of its interactions with various BH3-only proteins such as through posttranslational modifications. -

Identification of Potential Key Genes and Pathway Linked with Sporadic Creutzfeldt-Jakob Disease Based on Integrated Bioinformatics Analyses
medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Identification of potential key genes and pathway linked with sporadic Creutzfeldt-Jakob disease based on integrated bioinformatics analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.12.21.20248688; this version posted December 24, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Abstract Sporadic Creutzfeldt-Jakob disease (sCJD) is neurodegenerative disease also called prion disease linked with poor prognosis. The aim of the current study was to illuminate the underlying molecular mechanisms of sCJD. The mRNA microarray dataset GSE124571 was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened.