Supporting Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Localized Expression of a Myogenic Regulatory Gene, Qmfl, in the Somlte Dermatome of Avian Embryos
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Localized expression of a myogenic regulatory gene, qmfl, in the somlte dermatome of avian embryos Fabienne Charles de la Brousse and Charles P. Emerson, Jr. 1 Department of Biology, Gilmer Hall, University of Virginia, Charlottesville, Virginia 22901 USA qmfl is a quail myogenic regulatory gene that is transcribed in skeletal myoblasts and differentiated muscle and shows sequence homology to MyoD1 and MyfS. We used the qmfl transcript as an in situ hybridization marker for determined myogenic cells to study myogenic lineages in developing embryos. We present evidence for the temporal and spatial regulation of qmfl mRNA expression and slow cardiac troponin C (TnC), fast skeletal troponin T (TnT), and a-cardiac actin contractile protein mRNA expression in the somite myotome and limb buds. Our results show that qmfl is a marker for myogenic lineages during both somite formation and limb development and that qmfl mRNAs, but not contractile protein mRNAs, localize in dorsal medial lip (DML) cells of the somite dermatome. We propose that the DML is a site of myogenic lineage determination. [Key Words: qmfl; myogenesis; avian; somite; dermatome; myotome] Received December 1, 1989; revised version accepted January 25, 1990. Skeletal muscles of vertebrates are formed by the differ- compartmentalization process in which two and, subse- entiation of myoblasts in muscle-forming regions of the quently, three cell layers, including the sclerotome, der- embryo. In vitro, these determined myogenic cells pro- matome, and myotome, are formed. Ventromedial cells liferate clonally (Konigsberg 1963, 1971; Yaffe and Saxel of the sclerotome give rise to the connective tissue of 1977) and stably inherit the potential to differentiate the body and limb (Bancroft and Bellairs 1976; Solursh et into myofibers (Shainberg et al. -

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -
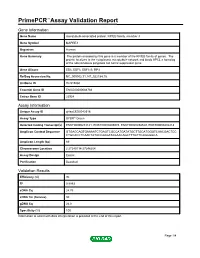
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name microtubule-associated protein, RP/EB family, member 3 Gene Symbol MAPRE3 Organism Human Gene Summary The protein encoded by this gene is a member of the RP/EB family of genes. The protein localizes to the cytoplasmic microtubule network and binds APCL a homolog of the adenomatous polyposis coli tumor suppressor gene. Gene Aliases EB3, EBF3, EBF3-S, RP3 RefSeq Accession No. NC_000002.11, NT_022184.15 UniGene ID Hs.515860 Ensembl Gene ID ENSG00000084764 Entrez Gene ID 22924 Assay Information Unique Assay ID qHsaCED0042518 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233121, ENST00000405074, ENST00000458529, ENST00000402218 Amplicon Context Sequence GTGACCAGTGAAAATCTGAGTCGCCATGATATGCTTGCATGGGTCAACGACTCC CTGCACCTCAACTATACCAAGATAGAACAGCTTTGTTCAGGGGCA Amplicon Length (bp) 69 Chromosome Location 2:27245114-27246204 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 90 R2 0.9993 cDNA Cq 24.76 cDNA Tm (Celsius) 80 gDNA Cq 26.8 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report MAPRE3, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Identification of Key Pathways and Genes in Endometrial Cancer Using Bioinformatics Analyses
ONCOLOGY LETTERS 17: 897-906, 2019 Identification of key pathways and genes in endometrial cancer using bioinformatics analyses YAN LIU, TENG HUA, SHUQI CHI and HONGBO WANG Department of Obstetrics and Gynecology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei 430022, P.R. China Received March 16, 2018; Accepted October 12, 2018 DOI: 10.3892/ol.2018.9667 Abstract. Endometrial cancer (EC) is one of the most Introduction common gynecological cancer types worldwide. However, to the best of our knowledge, its underlying mechanisms Endometrial carcinoma (EC) is one of the most common remain unknown. The current study downloaded three mRNA gynecological cancer types, with increasing global incidence and microRNA (miRNA) datasets of EC and normal tissue in recent years (1). A total of 60,050 cases of EC and 10,470 samples, GSE17025, GSE63678 and GSE35794, from the EC-associated cases of mortality were reported in the USA in Gene Expression Omnibus to identify differentially expressed 2016 (1), which was markedly higher than the 2012 statistics genes (DEGs) and miRNAs (DEMs) in EC tumor tissues. of 47,130 cases and 8,010 mortalities (2). Although numerous The DEGs and DEMs were then validated using data from studies have been conducted to investigate the mechanisms of The Cancer Genome Atlas and subjected to gene ontology endometrial tumorigenesis and development, to the best of our and Kyoto Encyclopedia of Genes and Genomes pathway knowledge, the exact etiology remains unknown. Understanding analysis. STRING and Cytoscape were used to construct a the potential molecular mechanisms underlying EC initiation protein-protein interaction network and the prognostic effects and progression is of great clinical significance. -

Seq2pathway Vignette
seq2pathway Vignette Bin Wang, Xinan Holly Yang, Arjun Kinstlick May 19, 2021 Contents 1 Abstract 1 2 Package Installation 2 3 runseq2pathway 2 4 Two main functions 3 4.1 seq2gene . .3 4.1.1 seq2gene flowchart . .3 4.1.2 runseq2gene inputs/parameters . .5 4.1.3 runseq2gene outputs . .8 4.2 gene2pathway . 10 4.2.1 gene2pathway flowchart . 11 4.2.2 gene2pathway test inputs/parameters . 11 4.2.3 gene2pathway test outputs . 12 5 Examples 13 5.1 ChIP-seq data analysis . 13 5.1.1 Map ChIP-seq enriched peaks to genes using runseq2gene .................... 13 5.1.2 Discover enriched GO terms using gene2pathway_test with gene scores . 15 5.1.3 Discover enriched GO terms using Fisher's Exact test without gene scores . 17 5.1.4 Add description for genes . 20 5.2 RNA-seq data analysis . 20 6 R environment session 23 1 Abstract Seq2pathway is a novel computational tool to analyze functional gene-sets (including signaling pathways) using variable next-generation sequencing data[1]. Integral to this tool are the \seq2gene" and \gene2pathway" components in series that infer a quantitative pathway-level profile for each sample. The seq2gene function assigns phenotype-associated significance of genomic regions to gene-level scores, where the significance could be p-values of SNPs or point mutations, protein-binding affinity, or transcriptional expression level. The seq2gene function has the feasibility to assign non-exon regions to a range of neighboring genes besides the nearest one, thus facilitating the study of functional non-coding elements[2]. Then the gene2pathway summarizes gene-level measurements to pathway-level scores, comparing the quantity of significance for gene members within a pathway with those outside a pathway. -

Human Transcription Factor Protein-Protein Interactions in Health and Disease
HELKA GÖÖS GÖÖS HELKA Recent Publications in this Series 45/2019 Mgbeahuruike Eunice Ego Evaluation of the Medicinal Uses and Antimicrobial Activity of Piper guineense (Schumach & Thonn) 46/2019 Suvi Koskinen AND DISEASE IN HEALTH INTERACTIONS PROTEIN-PROTEIN FACTOR HUMAN TRANSCRIPTION Near-Occlusive Atherosclerotic Carotid Artery Disease: Study with Computed Tomography Angiography 47/2019 Flavia Fontana DISSERTATIONES SCHOLAE DOCTORALIS AD SANITATEM INVESTIGANDAM Biohybrid Cloaked Nanovaccines for Cancer Immunotherapy UNIVERSITATIS HELSINKIENSIS 48/2019 Marie Mennesson Kainate Receptor Auxiliary Subunits Neto1 and Neto2 in Anxiety and Fear-Related Behaviors 49/2019 Zehua Liu Porous Silicon-Based On-Demand Nanohybrids for Biomedical Applications 50/2019 Veer Singh Marwah Strategies to Improve Standardization and Robustness of Toxicogenomics Data Analysis HELKA GÖÖS 51/2019 Iryna Hlushchenko Actin Regulation in Dendritic Spines: From Synaptic Plasticity to Animal Behavior and Human HUMAN TRANSCRIPTION FACTOR PROTEIN-PROTEIN Neurodevelopmental Disorders 52/2019 Heini Liimatta INTERACTIONS IN HEALTH AND DISEASE Efectiveness of Preventive Home Visits among Community-Dwelling Older People 53/2019 Helena Karppinen Older People´s Views Related to Their End of Life: Will-to-Live, Wellbeing and Functioning 54/2019 Jenni Laitila Elucidating Nebulin Expression and Function in Health and Disease 55/2019 Katarzyna Ciuba Regulation of Contractile Actin Structures in Non-Muscle Cells 56/2019 Sami Blom Spatial Characterisation of Prostate Cancer by Multiplex -

Pathway-Based Genome-Wide Association Analysis of Coronary Heart Disease Identifies Biologically Important Gene Sets
European Journal of Human Genetics (2012) 20, 1168–1173 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Pathway-based genome-wide association analysis of coronary heart disease identifies biologically important gene sets Lisa de las Fuentes1,4, Wei Yang2,4, Victor G Da´vila-Roma´n1 and C Charles Gu*,2,3 Genome-wide association (GWA) studies of complex diseases including coronary heart disease (CHD) challenge investigators attempting to identify relevant genetic variants among hundreds of thousands of markers being tested. A selection strategy based purely on statistical significance will result in many false negative findings after adjustment for multiple testing. Thus, an integrated analysis using information from the learned genetic pathways, molecular functions, and biological processes is desirable. In this study, we applied a customized method, variable set enrichment analysis (VSEA), to the Framingham Heart Study data (404 467 variants, n ¼ 6421) to evaluate enrichment of genetic association in 1395 gene sets for their contribution to CHD. We identified 25 gene sets with nominal Po0.01; at least four sets are previously known for their roles in CHD: vascular genesis (GO:0001570), fatty-acid biosynthetic process (GO:0006633), fatty-acid metabolic process (GO:0006631), and glycerolipid metabolic process (GO:0046486). Although the four gene sets include 170 genes, only three of the genes contain a variant ranked among the top 100 in single-variant association tests of the 404 467 variants tested. Significant enrichment for novel gene sets less known for their importance to CHD were also identified: Rac 1 cell-motility signaling pathway (h_rac1 Pathway, Po0.001) and sulfur amino-acid metabolic process (GO:0000096, Po0.001). -

Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers
Identification and Characterization of TPRKB Dependency in TP53 Deficient Cancers. by Kelly Kennaley A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Molecular and Cellular Pathology) in the University of Michigan 2019 Doctoral Committee: Associate Professor Zaneta Nikolovska-Coleska, Co-Chair Adjunct Associate Professor Scott A. Tomlins, Co-Chair Associate Professor Eric R. Fearon Associate Professor Alexey I. Nesvizhskii Kelly R. Kennaley [email protected] ORCID iD: 0000-0003-2439-9020 © Kelly R. Kennaley 2019 Acknowledgements I have immeasurable gratitude for the unwavering support and guidance I received throughout my dissertation. First and foremost, I would like to thank my thesis advisor and mentor Dr. Scott Tomlins for entrusting me with a challenging, interesting, and impactful project. He taught me how to drive a project forward through set-backs, ask the important questions, and always consider the impact of my work. I’m truly appreciative for his commitment to ensuring that I would get the most from my graduate education. I am also grateful to the many members of the Tomlins lab that made it the supportive, collaborative, and educational environment that it was. I would like to give special thanks to those I’ve worked closely with on this project, particularly Dr. Moloy Goswami for his mentorship, Lei Lucy Wang, Dr. Sumin Han, and undergraduate students Bhavneet Singh, Travis Weiss, and Myles Barlow. I am also grateful for the support of my thesis committee, Dr. Eric Fearon, Dr. Alexey Nesvizhskii, and my co-mentor Dr. Zaneta Nikolovska-Coleska, who have offered guidance and critical evaluation since project inception. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/)