Gene Duplication, Genome Duplication, and the Functional Diversification of Vertebrate Globins Jay F
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

"Evolutionary Emergence of Genes Through Retrotransposition"
Evolutionary Emergence of Advanced article Genes Through Article Contents . Introduction Retrotransposition . Gene Alteration Following Retrotransposon Insertion . Retrotransposon Recruitment by Host Genome . Retrotransposon-mediated Gene Duplication Richard Cordaux, University of Poitiers, Poitiers, France . Conclusion Mark A Batzer, Department of Biological Sciences, Louisiana State University, Baton Rouge, doi: 10.1002/9780470015902.a0020783 Louisiana, USA Variation in the number of genes among species indicates that new genes are continuously generated over evolutionary times. Evidence is accumulating that transposable elements, including retrotransposons (which account for about 90% of all transposable elements inserted in primate genomes), are potent mediators of new gene origination. Retrotransposons have fostered genetic innovation during human and primate evolution through: (i) alteration of structure and/or expression of pre-existing genes following their insertion, (ii) recruitment (or domestication) of their coding sequence by the host genome and (iii) their ability to mediate gene duplication via ectopic recombination, sequence transduction and gene retrotransposition. Introduction genes, respectively, and de novo origination from previ- ously noncoding genomic sequence. Genome sequencing Variation in the number of genes among species indicates projects have also highlighted that new gene structures can that new genes are continuously generated over evolution- arise as a result of the activity of transposable elements ary times. Although the emergence of new genes and (TEs), which are mobile genetic units or ‘jumping genes’ functions is of central importance to the evolution of that have been bombarding the genomes of most species species, studies on the formation of genetic innovations during evolution. For example, there are over three million have only recently become possible. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -
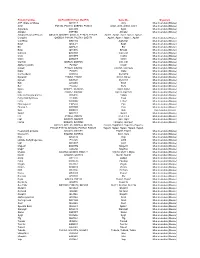
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures. -

Genome Organization/ Human
Genome Organization/ Secondary article Human Article Contents . Introduction David H Kass, Eastern Michigan University, Ypsilanti, Michigan, USA . Sequence Complexity Mark A Batzer, Louisiana State University Health Sciences Center, New Orleans, Louisiana, USA . Single-copy Sequences . Repetitive Sequences . The human nuclear genome is a highly complex arrangement of two sets of 23 Macrosatellites, Minisatellites and Microsatellites . chromosomes, or DNA molecules. There are various types of DNA sequences and Gene Families . chromosomal arrangements, including single-copy protein-encoding genes, repetitive Gene Superfamilies . sequences and spacer DNA. Transposable Elements . Pseudogenes . Mitochondrial Genome Introduction . Genome Evolution . Acknowledgements The human nuclear genome contains 3000 million base pairs (bp) of DNA, of which only an estimated 3% possess protein-encoding sequences. As shown in Figure 1, the DNA sequences of the eukaryotic genome can be classified sequences such as the ribosomal RNA genes. Repetitive into several types, including single-copy protein-encoding sequences with no known function include the various genes, DNA that is present in more than one copy highly repeated satellite families, and the dispersed, (repetitive sequences) and intergenic (spacer) DNA. The moderately repeated transposable element families. The most complex of these are the repetitive sequences, some of remainder of the genome consists of spacer DNA, which is which are functional and some of which are without simply a broad category of undefined DNA sequences. function. Functional repetitive sequences are classified into The human nuclear genome consists of 23 pairs of dispersed and/or tandemly repeated gene families that chromosomes, or 46 DNA molecules, of differing sizes either encode proteins (and may include noncoding (Table 1). -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -

Estimation of Duplication History Under a Stochastic Model for Tandem Repeats Farzad Farnoud1* , Moshe Schwartz2 and Jehoshua Bruck3
Farnoud et al. BMC Bioinformatics (2019) 20:64 https://doi.org/10.1186/s12859-019-2603-1 RESEARCH ARTICLE Open Access Estimation of duplication history under a stochastic model for tandem repeats Farzad Farnoud1* , Moshe Schwartz2 and Jehoshua Bruck3 Abstract Background: Tandem repeat sequences are common in the genomes of many organisms and are known to cause important phenomena such as gene silencing and rapid morphological changes. Due to the presence of multiple copies of the same pattern in tandem repeats and their high variability, they contain a wealth of information about the mutations that have led to their formation. The ability to extract this information can enhance our understanding of evolutionary mechanisms. Results: We present a stochastic model for the formation of tandem repeats via tandem duplication and substitution mutations. Based on the analysis of this model, we develop a method for estimating the relative mutation rates of duplications and substitutions, as well as the total number of mutations, in the history of a tandem repeat sequence. We validate our estimation method via Monte Carlo simulation and show that it outperforms the state-of-the-art algorithm for discovering the duplication history. We also apply our method to tandem repeat sequences in the human genome, where it demonstrates the different behaviors of micro- and mini-satellites and can be used to compare mutation rates across chromosomes. It is observed that chromosomes that exhibit the highest mutation activity in tandem repeat regions are the same as those thought to have the highest overall mutation rates. However, unlike previous works that rely on comparing human and chimpanzee genomes to measure mutation rates, the proposed method allows us to find chromosomes with the highest mutation activity based on a single genome, in essence by comparing (approximate) copies of the pattern in tandem repeats. -

Genomic Comparison of Closely Related Giant Viruses Supports an Accordion-Like Model of Evolution
ORIGINAL RESEARCH published: 16 June 2015 doi: 10.3389/fmicb.2015.00593 Genomic comparison of closely related Giant Viruses supports an accordion-like model of evolution Jonathan Filée * Laboratoire Evolution, Génome, Comportement, Ecologie, Centre National de la Recherche Scientifique UMR 9191, IRD UMR 247, Université Paris-Saclay, Gif-sur-Yvette, France Genome gigantism occurs so far in Phycodnaviridae and Mimiviridae (order Megavirales). Origin and evolution of these Giant Viruses (GVs) remain open questions. Interestingly, availability of a collection of closely related GV genomes enabling genomic comparisons offer the opportunity to better understand the different evolutionary forces acting on these genomes. Whole genome alignment for five groups of viruses belonging to the Mimiviridae and Phycodnaviridae families show that there is no trend of genome expansion or general tendency of genome contraction. Instead, GV genomes accumulated genomic mutations over the time with gene gains compensating the Edited by: different losses. In addition, each lineage displays specific patterns of genome evolution. Bernard La Scola, Mimiviridae (megaviruses and mimiviruses) and Chlorella Phycodnaviruses evolved Aix Marseille Université, France mainly by duplications and losses of genes belonging to large paralogous families Reviewed by: (including movements of diverse mobiles genetic elements), whereas Micromonas and Dahlene N. Fusco, Massachusetts General Hospital, USA Ostreococcus Phycodnaviruses derive most of their genetic novelties thought lateral Philippe Colson, gene transfers. Taken together, these data support an accordion-like model of evolution Aix-Marseille Université, France in which GV genomes have undergone successive steps of gene gain and gene loss, *Correspondence: Jonathan Filée, accrediting the hypothesis that genome gigantism appears early, before the diversification Laboratoire Evolution, Génome, of the different GV lineages. -

Extrachromosomal Element Capture and the Evolution of Multiple Replication Origins in Archaeal Chromosomes
Extrachromosomal element capture and the evolution of multiple replication origins in archaeal chromosomes Nicholas P. Robinson† and Stephen D. Bell† Medical Research Council Cancer Cell Unit, Hutchison Medical Research Council Research Center, Hills Road, Cambridge CB2 0XZ, United Kingdom Edited by Carl R. Woese, University of Illinois at Urbana–Champaign, Urbana, IL, and approved February 15, 2007 (received for review January 9, 2007) In all three domains of life, DNA replication begins at specialized Orc2–6 act to recruit MCM to origins of replication in a reaction loci termed replication origins. In bacteria, replication initiates from that absolutely requires an additional factor, Cdt1 (6). Although a single, clearly defined site. In contrast, eukaryotic organisms archaea possess orthologs of Orc1, Cdc6, and MCM, no archaeal exploit a multitude of replication origins, dividing their genomes homolog of Cdt1 has yet been identified. into an array of short contiguous units. Recently, the multiple In the current work, we reveal that Aeropyrum pernix has at replication origin paradigm has also been demonstrated within the least two replication origins, indicating that the multiple repli- archaeal domain of life, with the discovery that the hyperthermo- cation origin paradigm is not restricted to the Sulfolobus genus. philic archaeon Sulfolobus has three replication origins. However, Comparison of the A. pernix and Sulfolobus origins reveals a clear the evolutionary mechanism driving the progression from single to relationship between these loci. Further, analyses of the gene multiple origin usage remains unclear. Here, we demonstrate that order and identity in the environment of the origins provides Aeropyrum pernix, a distant relative of Sulfolobus, has two origins. -

Mitochondrial DNA Duplication, Recombination, and Introgression During Interspecific Hybridization
www.nature.com/scientificreports OPEN Mitochondrial DNA duplication, recombination, and introgression during interspecifc hybridization Silvia Bágeľová Poláková1,5, Žaneta Lichtner1, Tomáš Szemes2,3,4, Martina Smolejová1 & Pavol Sulo1* mtDNA recombination events in yeasts are known, but altered mitochondrial genomes were not completed. Therefore, we analyzed recombined mtDNAs in six Saccharomyces cerevisiae × Saccharomyces paradoxus hybrids in detail. Assembled molecules contain mostly segments with variable length introgressed to other mtDNA. All recombination sites are in the vicinity of the mobile elements, introns in cox1, cob genes and free standing ORF1, ORF4. The transplaced regions involve co-converted proximal exon regions. Thus, these selfsh elements are benefcial to the host if the mother molecule is challenged with another molecule for transmission to the progeny. They trigger mtDNA recombination ensuring the transfer of adjacent regions, into the progeny of recombinant molecules. The recombination of the large segments may result in mitotically stable duplication of several genes. Hybrids between Saccharomyces species occur frequently in nature as a number of hybrids have been reported among wine and beer strains1–6. Most lager beer strains are hybrids between S. cerevisiae and S. eubayanus, combining the ability to produce ethanol with cryotolerance 6–8. Some S. cerevisiae × S. kudriavzevii strains are associated with beer, but most of them are associated with wine, where they provide unique favor8,9. S. eubayanus and S. uvarum hybrids have been associated with sparkling wine, cider fermentation, and, in some cases, with the production of of-favors in breweries 8,10,11. Interspecifc hybrids among Saccharomyces species can also be readily obtained in the laboratory 9,12–14.