An Overview of Duplicated Gene Detection Methods: Why the Duplication Mechanism Has to Be Accounted for in Their Choice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Expression of a Mouse Long Terminal Repeat Is Cell Cycle-Linked (Friend Cells/Gene Expression/Retroviruses/Transformation/Onc Genes) LEONARD H
Proc. Natl. Acad. Sci. USA Vol. 82, pp. 1946-1949, April 1985 Biochemistry Expression of a mouse long terminal repeat is cell cycle-linked (Friend cells/gene expression/retroviruses/transformation/onc genes) LEONARD H. AUGENLICHT AND HEIDI HALSEY Department of Oncology, Montefiore Medical Center, and Department of Medicine, Albert Einstein College of Medicine, 111 East 210th St., Bronx, NY 10467 Communicated by Harry Eagle, November 26, 1984 ABSTRACT The expression of the long terminal repeat have regions of homology with a Syrian hamster repetitive (LTR) of intracisternal A particle retroviral sequences which sequence whose expression is also linked to the G, phase of are endogenous to the mouse genome has been shown to be the cell cycle (28). linked to the early G6 phase of the cell cycle in Friend erythroleukemia cells synchronized by density arrest and also MATERIALS AND METHODS in logarithmically growing cells fractionated into cell-cycle Cells. Friend erythroleukemia cells, strain DS-19, were compartments by centrifugal elutriation. Regions of homology grown in minimal essential medium containing 10% fetal calf were found in comparing the LTR sequence to a repetitive serum (28). Cell number was determined by counting an Syrian hamster sequence specifically expressed in early G1 in aliquot in an automated particle counter (Coulter Electron- hamster cells. ics). The cells were fractionated into cell-cycle compart- ments by centrifugal elutriation with a Beckman elutriator The long terminal repeats (LTR) of retroviral genomes rotor as described (29). For analysis of DNA content per contain sequence elements that regulate the transcription of cell, the cells were stained with either 4',6-diamidino-2- the viral genes (1). -

Best Practice Recommendations for Internal Validation of Human Short Tandem Repeat Profiling on Capillary Electrophoresis Platforms
Best Practice Recommendations for Internal Validation of Human Short Tandem Repeat Profiling on Capillary Electrophoresis Platforms Best Practice Recommendations for Internal Validation of Human Short Tandem Repeat Profiling on Capillary Electrophoresis Platforms Biological Methods Subcommittee Biology/DNA Scientific Area Committee Organization of Scientific Area Committees (OSAC) for Forensic Science Best Practice Recommendations for Internal Validation of Human Short Tandem Repeat Profiling on Capillary Electrophoresis Platforms OSAC Proposed Standard Best Practice Recommendations for Internal Validation of Human Short Tandem Repeat Profiling on Capillary Electrophoresis Platforms Prepared by Biological Methods Subcommittee Version: 1.0 Disclaimer: This document has been developed by the Biological Methods Subcommittee of the Organization of Scientific Area Committees (OSAC) for Forensic Science through a consensus process and is proposed for further development through a Standard Developing Organization (SDO). This document is being made available so that the forensic science community and interested parties can consider the recommendations of the OSAC pertaining to applicable forensic science practices. The document was developed with input from experts in a broad array of forensic science disciplines as well as scientific research, measurement science, statistics, law, and policy. This document has not been published by an SDO. Its contents are subject to change during the standards development process. All interested groups or individuals are strongly encouraged to submit comments on this proposed document during the open comment period administered by the Academy Standards Board (www.asbstandardsboard.org). Best Practice Recommendations for Internal Validation of Human Short Tandem Repeat Profiling on Capillary Electrophoresis Platforms Foreword This document outlines best practice recommendations for the internal validation of human short tandem repeat DNA profiling on capillary electrophoresis platforms utilized in forensic laboratories. -

"Evolutionary Emergence of Genes Through Retrotransposition"
Evolutionary Emergence of Advanced article Genes Through Article Contents . Introduction Retrotransposition . Gene Alteration Following Retrotransposon Insertion . Retrotransposon Recruitment by Host Genome . Retrotransposon-mediated Gene Duplication Richard Cordaux, University of Poitiers, Poitiers, France . Conclusion Mark A Batzer, Department of Biological Sciences, Louisiana State University, Baton Rouge, doi: 10.1002/9780470015902.a0020783 Louisiana, USA Variation in the number of genes among species indicates that new genes are continuously generated over evolutionary times. Evidence is accumulating that transposable elements, including retrotransposons (which account for about 90% of all transposable elements inserted in primate genomes), are potent mediators of new gene origination. Retrotransposons have fostered genetic innovation during human and primate evolution through: (i) alteration of structure and/or expression of pre-existing genes following their insertion, (ii) recruitment (or domestication) of their coding sequence by the host genome and (iii) their ability to mediate gene duplication via ectopic recombination, sequence transduction and gene retrotransposition. Introduction genes, respectively, and de novo origination from previ- ously noncoding genomic sequence. Genome sequencing Variation in the number of genes among species indicates projects have also highlighted that new gene structures can that new genes are continuously generated over evolution- arise as a result of the activity of transposable elements ary times. Although the emergence of new genes and (TEs), which are mobile genetic units or ‘jumping genes’ functions is of central importance to the evolution of that have been bombarding the genomes of most species species, studies on the formation of genetic innovations during evolution. For example, there are over three million have only recently become possible. -

RESEARCH ARTICLES Gene Cluster Statistics with Gene Families
RESEARCH ARTICLES Gene Cluster Statistics with Gene Families Narayanan Raghupathy*1 and Dannie Durand* *Department of Biological Sciences, Carnegie Mellon University, Pittsburgh, PA; and Department of Computer Science, Carnegie Mellon University, Pittsburgh, PA Identifying genomic regions that descended from a common ancestor is important for understanding the function and evolution of genomes. In distantly related genomes, clusters of homologous gene pairs are evidence of candidate homologous regions. Demonstrating the statistical significance of such ‘‘gene clusters’’ is an essential component of comparative genomic analyses. However, currently there are no practical statistical tests for gene clusters that model the influence of the number of homologs in each gene family on cluster significance. In this work, we demonstrate empirically that failure to incorporate gene family size in gene cluster statistics results in overestimation of significance, leading to incorrect conclusions. We further present novel analytical methods for estimating gene cluster significance that take gene family size into account. Our methods do not require complete genome data and are suitable for testing individual clusters found in local regions, such as contigs in an unfinished assembly. We consider pairs of regions drawn from the same genome (paralogous clusters), as well as regions drawn from two different genomes (orthologous clusters). Determining cluster significance under general models of gene family size is computationally intractable. By assuming that all gene families are of equal size, we obtain analytical expressions that allow fast approximation of cluster probabilities. We evaluate the accuracy of this approximation by comparing the resulting gene cluster probabilities with cluster probabilities obtained by simulating a realistic, power-law distributed model of gene family size, with parameters inferred from genomic data. -
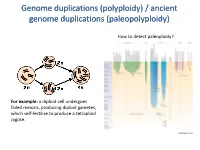
Polyploidy) / Ancient Genome Duplications (Paleopolyploidy
Genome duplications (polyploidy) / ancient genome duplications (paleopolyploidy) How to detect paleoploidy? For example: a diploid cell undergoes failed meiosis, producing diploid gametes, which self-fertilize to produce a tetraploid zygote. Timing of duplication by trees (phylogenetic timing) Phylogenetic timing of duplicates b Paramecium genome duplications Comparison of two scaffolds originating from a common ancestor at the recent WGD Saccharomyces cerevisiae Just before genome duplication Just after genome duplication More time after genome duplication Unaligned view (removing gaps just like in cerev has occurred) Saccharomyces cerevisiae Problem reciprocal gene loss (extreme case); how to solve? Problem reciprocal gene loss (extreme case); how to solve? Just before genome duplication Outgroup! Just after genome duplication Outgroup Just after genome duplication Outgroup More time after genome duplication Outgroup Problem (extreme case); how to solve? Outgroup Outgroup Outgroup Outgroup Outgroup Using other genomes Wong et al. 2002 PNAS Centromeres Vertebrate genome duplication Nature. 2011 Apr 10. [Epub ahead of print] Ancestral polyploidy in seed plants and angiosperms. Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, Depamphilis CW. Flowering plants Flowering MOSS Vertebrates Teleosts S. serevisiae and close relatives Paramecium Reconstructed map of genome duplications allows unprecedented mapping -

A 'David and Goliath'
Current Genomics, 2002, 3, 563-576 563 Nucleolar Dominance: A ‘David and Goliath’ Chromatin Imprinting Process W. Viegas1,*, N. Neves1,2, M. Silva1, A. Caperta1,2, and L. Morais-Cecílio1 1Centro de Botânica Aplicada à Agricultura, Secção de Genética/DBEB, Instituto Superior de Agronomia, 1349-017 Lisboa, 2Departamento de Ciências Biológicas e Naturais, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande 376, 1749-042 Lisboa, Portugal Abstract: Nucleolar dominance is an enigma. The puzzle of differential amphiplasty has remained unresolved since it was first recognised and described in Crepis hybrids by Navashin in 1934. Here we review the body of knowledge that has grown out of the many models that have tried to find the genetic basis for differential rRNA gene expression in hybrids, and present a new interpretation. We propose and discuss a chromatin imprinting model which re-interprets differential amphiplasty in terms of two genomes of differing size occupying a common space within the nucleus, and with heterochromatin as a key player in the scenario. Difference in size between two parental genomes induces an inherited epigenetic mark in the hybrid that allows patterns of chromatin organization to have positional effects on the neighbouring domains. This chromatin imprinting model can be also used to explain complex genomic interactions which transcend nucleolar dominance and which can account for the overall characteristics of hybrids. Gene expression in hybrids, relative to parentage, is seen as being based on the nuclear location of the sequences concerned within their genomic environment, and where the presence of particular repetitive DNA sequences are ‘sensed’, and render silent the adjacent information. -

Microsatellites Within the Feline Androgen Receptor Are
JFM0010.1177/1098612X16634386Journal of Feline Medicine and SurgeryFarwick et al 634386research-article2016 Original Article Journal of Feline Medicine and Surgery 1 –7 Microsatellites within the feline © The Author(s) 2016 Reprints and permissions: androgen receptor are suitable for X sagepub.co.uk/journalsPermissions.nav DOI: 10.1177/1098612X16634386 chromosome-linked clonality testing jfms.com in archival material Nadine M Farwick1, Robert Klopfleisch1, Achim D Gruber1 and Alexander Th A Weiss2 Abstract Objectives A hallmark of neoplasms is their origin from a single cell; that is, clonality. Many techniques have been developed in human medicine to utilise this feature of tumours for diagnostic purposes. One approach is X chromosome-linked clonality testing using polymorphisms of genes encoded by genes on the X chromosome. The aim of this study was to determine if the feline androgen receptor gene was suitable for X chromosome-linked clonality testing. Methods The feline androgen receptor gene, was characterised and used to test clonality of feline lymphomas by PCR and polyacrylamide gel electrophoresis, using archival formalin-fixed, paraffin-embedded material. Results Clonality of the feline lymphomas under study was confirmed and the gene locus was shown to represent a suitable target in clonality testing. Conclusions and relevance Because there are some pitfalls using X chromosome-linked clonality testing, further studies are necessary to establish this technique in the cat. Accepted: 26 January 2016 Introduction Most tumours arise -

(Lcrs) in 22Q11 Mediate Deletions, Duplications, Translocations, and Genomic Instability: an Update and Literature Review Tamim H
review January/February 2001 ⅐ Vol. 3 ⅐ No. 1 Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: An update and literature review Tamim H. Shaikh, PhD1, Hiroki Kurahashi, MD, PhD1, and Beverly S. Emanuel, PhD1,2 Several constitutional rearrangements, including deletions, duplications, and translocations, are associated with 22q11.2. These rearrangements give rise to a variety of genomic disorders, including DiGeorge, velocardiofacial, and conotruncal anomaly face syndromes (DGS/VCFS/CAFS), cat eye syndrome (CES), and the supernumerary der(22)t(11;22) syndrome associated with the recurrent t(11;22). Chromosome 22-specific duplications or low copy repeats (LCRs) have been directly implicated in the chromosomal rearrangements associated with 22q11.2. Extensive sequence analysis of the different copies of 22q11 LCRs suggests a complex organization. Examination of their evolutionary origin suggests that the duplications in 22q11.2 may predate the divergence of New World monkeys 40 million years ago. Based on the current data, a number of models are proposed to explain the LCR-mediated constitutional rearrangements of 22q11.2. Genetics in Medicine, 2001:3(1):6–13. Key Words: duplication, evolution, 22q11, deletion and translocation Although chromosome 22 represents only 2% of the haploid The 22q11.2 deletion syndrome, which includes DGS/ human genome,1 recurrent, clinically significant, acquired, VCFS/CAFS, is the most common microdeletion syndrome. and somatic -

A Field Guide to Eukaryotic Transposable Elements
GE54CH23_Feschotte ARjats.cls September 12, 2020 7:34 Annual Review of Genetics A Field Guide to Eukaryotic Transposable Elements Jonathan N. Wells and Cédric Feschotte Department of Molecular Biology and Genetics, Cornell University, Ithaca, New York 14850; email: [email protected], [email protected] Annu. Rev. Genet. 2020. 54:23.1–23.23 Keywords The Annual Review of Genetics is online at transposons, retrotransposons, transposition mechanisms, transposable genet.annualreviews.org element origins, genome evolution https://doi.org/10.1146/annurev-genet-040620- 022145 Abstract Annu. Rev. Genet. 2020.54. Downloaded from www.annualreviews.org Access provided by Cornell University on 09/26/20. For personal use only. Copyright © 2020 by Annual Reviews. Transposable elements (TEs) are mobile DNA sequences that propagate All rights reserved within genomes. Through diverse invasion strategies, TEs have come to oc- cupy a substantial fraction of nearly all eukaryotic genomes, and they rep- resent a major source of genetic variation and novelty. Here we review the defining features of each major group of eukaryotic TEs and explore their evolutionary origins and relationships. We discuss how the unique biology of different TEs influences their propagation and distribution within and across genomes. Environmental and genetic factors acting at the level of the host species further modulate the activity, diversification, and fate of TEs, producing the dramatic variation in TE content observed across eukaryotes. We argue that cataloging TE diversity and dissecting the idiosyncratic be- havior of individual elements are crucial to expanding our comprehension of their impact on the biology of genomes and the evolution of species. 23.1 Review in Advance first posted on , September 21, 2020. -

2R Or Not 2R Is Not the Question Anymore
CORRESPONDENCE LINK TO ORIGINAL ARTICLE LINK TO INITIAL CORRESPONDENCE origin of evolutionary novelties are highly interesting questions, but they remain 2R or not 2R is not the largely unsolved11. However, evidence for the two rounds of genome duplication dur- question anymore ing chordate evolution is very strong, and it would seem safe to say that the debate Yves Van de Peer, Steven Maere and Axel Meyer over 2R is settled and is no longer an open question12. As we point out in our Opinion article1, it is only the evolutionary effects of In his comments on our Opinion article in passing in our recent review on WGDs events such as WGDs on evolution that are (The evolutionary significance of ancient and their significance for evolution in Nature debated1,11, and no longer whether or not genome duplications. Nature Rev. Genet. Reviews Genetics1. two rounds of WGD occurred during the 10, 725–732 (2009))1 Amir Ali Abbasi Abbasi2, however, questions the support evolution of chordates. (Piecemeal or big bangs: correlating the for the 2R hypothesis, claiming that it still Yves Van de Peer and Steven Maere are at the vertebrate evolution with proposed models remains debated today. He points out cor- Department of Plant Systems Biology, VIB (Flanders of gene expansion events. Nature Rev. Genet. rectly that evidence for the 2R hypothesis Institute of Biotechnology), B-9052 Ghent, Belgium. 6 Jan 2010 (doi:10.1038/nrg2600-c1))2 was based initially on data from only a Axel Meyer is at the Department of Biology, University argues that it is not justified to speculate small number of genes and vertebrate and of Konstanz, D-78457 Konstanz, Germany. -

B2 and ALU Retrotransposons Are Self-Cleaving Ribozymes Whose Activity Is Enhanced by EZH2
B2 and ALU retrotransposons are self-cleaving ribozymes whose activity is enhanced by EZH2 Alfredo J. Hernandeza,1, Athanasios Zovoilisb,c,1,2, Catherine Cifuentes-Rojasb,c,1,3, Lu Hanb,c, Bojan Bujisicb,c, and Jeannie T. Leeb,c,4 aDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115; bDepartment of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114; and cDepartment of Genetics, The Blavatnik Institute, Harvard Medical School, Boston, MA 02114 Contributed by Jeannie T. Lee, November 18, 2019 (sent for review October 9, 2019; reviewed by Vivian G. Cheung and Karissa Y. Sanbonmatsu) Transposable elements make up half of the mammalian genome. B2s are present in ∼350,000 copies in the mouse genome (10) One of the most abundant is the short interspersed nuclear and give rise to 180- to 200-nucleotide (nt) polymerase III complex element (SINE). Among their million copies, B2 accounts for (POL-III) transcripts. B2 SINEs are transcriptionally active, with ∼350,000 in the mouse genome and has garnered special interest expression being highest in germ cells and in early development, because of emerging roles in epigenetic regulation. Our recent work after which they become epigenetically silenced. However, one of demonstrated that B2 RNA binds stress genes to retard transcription the most intriguing aspects of SINE biology is that these elements elongation. Although epigenetically silenced, B2s become massively become highly up-regulated when somatic cells are acutely stressed up-regulated during thermal and other types of stress. Specifically, (11–13), including during viral infection (8, 11), autoimmune pro- an interaction between B2 RNA and the Polycomb protein, EZH2, cesses such as macular degeneration (14, 15), and progression to results in cleavage of B2 RNA, release of B2 RNA from chromatin, cancer (16, 17). -

Tandemly Arrayed Genes in Vertebrate Genomes
Tandemly Arrayed Genes in Vertebrate Genomes Deng Pan1 and Liqing Zhang2∗ 1Department of Computer Science, Virginia Tech, 2050 Torgerson Hall, Blacksburg, VA 24061-0106, USA 2Program in Genetics, Bioinformatics, and Computational Biology ∗To whom correspondence should be addressed; E-mail: [email protected] July 9, 2008 Abstract Tandemly arrayed genes (TAGs) are duplicated genes that are linked as neighbors on a chromosome, many of which have important physio- logical and biochemical functions. Here we performed a survey of these genes in 11 available vertebrate genomes. TAGs account for an average of about 14% of all genes in these vertebrate genomes, and about 25% of all duplications. The majority of TAGs (72-94%) have parallel tran- scription orientation (i.e. are encoded on the same strand), in contrast to the genome, which has about 50% of its genes in parallel transcrip- tion orientation. The majority of tandem arrays have only two members. In all species, the proportion of genes that belong to TAGs tends to be higher in large gene families than in small ones; together with our re- cent finding that tandem duplication played a more important role than retroposition in large families, this fact suggests that among all types of duplication mechanisms, tandem duplication is the predominant mecha- nism of duplication, especially in large families. Species-specific tandem arrays (SSTAs) are identified and results of statistical tests suggest that natural selection played a role in maintaining some of the SSTAs. Our 1 work will provide more insight into the evolutionary history of TAGs in vertebrates. 2 1 Introduction Although the importance of duplicated genes in providing raw materials for genetic innovation has been recognized since the 1930s and is highlighted in Ohno’s book Evolution by gene duplication (Ohno, 1970), it is only recently that the availability of numerous genomic sequences has made it possible to quantitatively estimate how many genes in a genome are generated by gene duplication.