Gene Loss and Adaptation in Saccharomyces Genomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
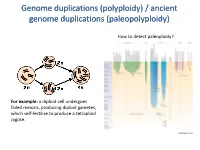
Polyploidy) / Ancient Genome Duplications (Paleopolyploidy
Genome duplications (polyploidy) / ancient genome duplications (paleopolyploidy) How to detect paleoploidy? For example: a diploid cell undergoes failed meiosis, producing diploid gametes, which self-fertilize to produce a tetraploid zygote. Timing of duplication by trees (phylogenetic timing) Phylogenetic timing of duplicates b Paramecium genome duplications Comparison of two scaffolds originating from a common ancestor at the recent WGD Saccharomyces cerevisiae Just before genome duplication Just after genome duplication More time after genome duplication Unaligned view (removing gaps just like in cerev has occurred) Saccharomyces cerevisiae Problem reciprocal gene loss (extreme case); how to solve? Problem reciprocal gene loss (extreme case); how to solve? Just before genome duplication Outgroup! Just after genome duplication Outgroup Just after genome duplication Outgroup More time after genome duplication Outgroup Problem (extreme case); how to solve? Outgroup Outgroup Outgroup Outgroup Outgroup Using other genomes Wong et al. 2002 PNAS Centromeres Vertebrate genome duplication Nature. 2011 Apr 10. [Epub ahead of print] Ancestral polyploidy in seed plants and angiosperms. Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, Depamphilis CW. Flowering plants Flowering MOSS Vertebrates Teleosts S. serevisiae and close relatives Paramecium Reconstructed map of genome duplications allows unprecedented mapping -

Copying out Our Abcs the Role of Gene Redundancy in Interpreting Genetic Hierarchies
Genomic imprinting in mammals COMMENT Outlook 14 Nicholls, R.D. et al. (1998) Imprinting in Prader–Willi and 21 Feil, R. et al. (1997) Parental chromosome-specific chromatin 28 Macleod, D. et al. (1994) Sp1 sites in the mouse Aprt gene Angelman syndromes. Trends Genet. 14, 194–199 conformation in the imprinted U2af1-rs1 gene in the mouse. promoter are required to prevent methylation of the CpG 15 Hark, A.T. and Tilghman, S.M. (1998) Chromatin conformation J. Biol. Chem. 272, 20893–20900 island. Genes Dev. 8, 2282–2292 of the H19 epigenetic mark. Hum. Mol. Genet. 7, 1979–1985 22 Schweizer, J. et al. (1999) In vivo nuclease hypersensitivity 29 Brandeis, M. et al. (1994) SP1 elements protect a CpG island 16 Szabó, P.E. et al. (1998) Characterization of novel parent- studies reveal multiple sites of parental-origin-dependent from de novo methylation. Nature 371, 435–438 specific epigenetic modifications upstream of the imprinted differential chromatin conformation in the 150 kb SNRPN 30 Kirillov, A. et al. (1996) A role for nuclear NF-kB in mouse H19 gene. Mol. Cell. Biol. 18, 6767–6776 transcription unit. Hum. Mol. Genet. 8, 555–566 B-cell-specific demethylation of the Igk locus. Nat. Genet. 13, 17 Khosla, S. et al. (1999) Parental allele-specific chromatin 23 Lyko, F. et al. (1998) Identification of a silencing element in 435–441 configuration in a boundary/imprinting-control element the human 15q11–q13 imprinting center by using 31 Hsieh, C-L. (1999) Evidence that protein-binding upstream of the mouse H19 gene. -

A 'David and Goliath'
Current Genomics, 2002, 3, 563-576 563 Nucleolar Dominance: A ‘David and Goliath’ Chromatin Imprinting Process W. Viegas1,*, N. Neves1,2, M. Silva1, A. Caperta1,2, and L. Morais-Cecílio1 1Centro de Botânica Aplicada à Agricultura, Secção de Genética/DBEB, Instituto Superior de Agronomia, 1349-017 Lisboa, 2Departamento de Ciências Biológicas e Naturais, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande 376, 1749-042 Lisboa, Portugal Abstract: Nucleolar dominance is an enigma. The puzzle of differential amphiplasty has remained unresolved since it was first recognised and described in Crepis hybrids by Navashin in 1934. Here we review the body of knowledge that has grown out of the many models that have tried to find the genetic basis for differential rRNA gene expression in hybrids, and present a new interpretation. We propose and discuss a chromatin imprinting model which re-interprets differential amphiplasty in terms of two genomes of differing size occupying a common space within the nucleus, and with heterochromatin as a key player in the scenario. Difference in size between two parental genomes induces an inherited epigenetic mark in the hybrid that allows patterns of chromatin organization to have positional effects on the neighbouring domains. This chromatin imprinting model can be also used to explain complex genomic interactions which transcend nucleolar dominance and which can account for the overall characteristics of hybrids. Gene expression in hybrids, relative to parentage, is seen as being based on the nuclear location of the sequences concerned within their genomic environment, and where the presence of particular repetitive DNA sequences are ‘sensed’, and render silent the adjacent information. -

Iv Gene Duplication and the Evolution of Silenced
Gene Duplication and the Evolution of Silenced Chromatin in Yeasts by Meleah A. Hickman University Program in Genetics and Genomics Duke University Date:_______________________ Approved: ___________________________ Laura Rusche, Advisor ___________________________ Paul Magwene, Chair ___________________________ Fred Dietrich ___________________________ Joe Heitman ___________________________ Beth Sullivan Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2010 i v ABSTRACT Gene Duplication and the Evolution of Silenced Chromatin in Yeasts by Meleah A. Hickman University Program in Genetics and Genomics Duke University Date:_______________________ Approved: ___________________________ Laura Rusche, Advisor ___________________________ Paul Magwene, Chair ___________________________ Fred Dietrich ___________________________ Joe Heitman ___________________________ Beth Sullivan An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2010 Copyright by Meleah A. Hickman 2010 Abstract In Saccharomyces cerevisiae, proper maintenance of haploid cell identity requires the SIR complex to mediate the silenced chromatin found at the cryptic mating-type loci, HML and HMR. This complex consists of Sir2, a histone deacetylase, and the histone binding proteins Sir3 and Sir4. Interestingly, both Sir2 and Sir3 have paralogs from a genome duplication that occurred after the divergence of Saccharomyces and Kluyveromyces species, approximately 100 million years ago. The histone deacetylase HST1 is the paralog of SIR2 and works with the promoter-specific SUM1 complex to repress sporulation and alpha-specific genes. ORC1 is the paralog of SIR3 and is an essential subunit of the Origin Recognition Complex and also recruits SIR proteins to the HM loci. -

Whole-Genome Duplications and Paleopolyploidy Whole-Genome Duplications
Whole-genome duplications and paleopolyploidy Whole-genome duplications AUTOPOLYPLOIDY ALLOPOLYPLOIDY Hegarty and HiscockHegarty 2008, 18 Current Biology Examples of allopolyploid speciation Hegarty and Hiscock 2008, Current Biology 18 Whole-genome duplications of different age time neopolyploidy mesopolyploidy paleopolyploidy time Whole-genome duplications in protozoa •Aury et al. (2006) analyzed the unicellular eukaryote Paramecium tetraurelia • most of 40,000 genes arose through at least 3 successive whole-genome duplications (WGDs) • most recent duplication most likely caused an explosion of speciation events that gave rise to the P. aurelia complex (15 sibling species) • some genes have been lost, some retained • many retained (duplicated) genes do not generate functional innovations but are important because of the gene dosage effect Whole-genome duplications in yeast • genome comparison between two yeast species, Saccharomyces cerevisiae (n=16) and Kluyveromyces waltii (n=8) • each region of K. waltii corresponding to two regions of S. cerevisiae •the S. cerevisiae genome underwent a WGD after the two yeast species diverged • in nearly every case (95%), accelerated evolution was confined to only one of the two paralogues (= one of the paralogues retained an ancestral function, the other was free to evolve more rapidly and acquired a derived function) Kellis et al. 2004, Nature 428 Whole-genome duplications in yeast Kellis et al. 2004, Nature 428 a) after divergence from K. waltii, the Saccharomyces lineage underwent a genome duplication event (2 copies of every gene and chromosome) b) duplicated genes were mutated and some lost c) two copies kept for only a small minority of duplicated genes d) the conserved order of duplicated genes (nos. -

Transposable Elements in Polyploid Evolution Final.Pdf
Transposable elements and polyploid evolution in animals Fernando Rodriguez and Irina R. Arkhipova Address Josephine Bay Paul Center for Comparative Molecular Biology and Evolution, Marine Biological Laboratory, Woods Hole, MA 02543, USA Corresponding author: Arkhipova, Irina R. ([email protected]) Polyploidy in animals is much less common than in plants, where it is thought to be pervasive in all higher plant lineages. Recent studies have highlighted the impact of polyploidization and the associated process of diploidy restoration on the evolution and speciation of selected taxonomic groups in the animal kingdom: from vertebrates represented by salmonid fishes and African clawed frogs to invertebrates represented by parasitic root-knot nematodes and bdelloid rotifers. In this review, we focus on the unique and diverse roles that transposable elements may play in these processes, from marking and diversifying subgenome-specific chromosome sets prior to hybridization, to influencing genome restructuring during rediploidization, to affecting subgenome-specific regulatory evolution, and occasionally providing opportunities for domestication and gene amplification to restore and improve functionality. There is still much to be learned from the future comparative genomic studies of chromosome-sized and haplotype-aware assemblies, and from post-genomic studies elucidating genetic and epigenetic regulatory phenomena across short and long evolutionary distances in the metazoan tree of life. Glossary Polyploidy: having more than the usual diploid set of homologous chromosomes Allopolyploidy: a form of polyploidy that results from interspecific hybridization Autopolyploidy: a form of polyploidy that results from chromosome redoubling in the same species Paleopolyploidy: ancient polyploidy Neopolyploidy: recent polyploidy Homeologs: pairs of genes originated by speciation and brought back together by allopolyploidization Ohnologs: same as homeologs, named after S. -

The Chromosome-Based Lavender Genome Provides New Insights Into
Li et al. Horticulture Research (2021) 8:53 Horticulture Research https://doi.org/10.1038/s41438-021-00490-6 www.nature.com/hortres ARTICLE Open Access The chromosome-based lavender genome provides new insights into Lamiaceae evolution and terpenoid biosynthesis Jingrui Li1,2, Yiming Wang 3,YanmeiDong1,2, Wenying Zhang1,2,DiWang1, Hongtong Bai1,KuiLi3,HuiLi1 and Lei Shi1 Abstract The aromatic shrub Lavandula angustifolia produces various volatile terpenoids that serve as resources for essential oils and function in plant-insect communication. To better understand the genetic basis of the terpenoid diversity in lavender, we present a high-quality reference genome for the Chinese lavender cultivar ‘Jingxun 2’ using PacBio and Hi-C technologies to anchor the 894.50 Mb genome assembly into 27 pseudochromosomes. In addition to the γ triplication event, lavender underwent two rounds of whole-genome duplication (WGD) during the Eocene–Oligocene (29.6 MYA) and Miocene–Pliocene (6.9 MYA) transitions. As a result of tandem duplications and lineage-specific WGDs, gene families related to terpenoid biosynthesis in lavender are substantially expanded compared to those of five other species in Lamiaceae. Many terpenoid biosynthesis transcripts are abundant in glandular trichomes. We further integrated the contents of ecologically functional terpenoids and coexpressed terpenoid biosynthetic genes to construct terpenoid-gene networks. Typical gene clusters, including TPS-TPS, TPS- CYP450, and TPS-BAHD, linked with compounds that primarily function as attractants or repellents, were identified by fl 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; their similar patterns of change during ower development or in response to methyl jasmonate. Comprehensive analysis of the genetic basis of the production of volatiles in lavender could serve as a foundation for future research into lavender evolution, phytochemistry, and ecology. -

Genetic Variation of the Serine Acetyltransferase Gene Family for Sulfur Assimilation in Maize
G C A T T A C G G C A T genes Article Genetic Variation of the Serine Acetyltransferase Gene Family for Sulfur Assimilation in Maize Zhixuan Zhao 1, Shuai Li 1, Chen Ji 2 , Yong Zhou 2, Changsheng Li 2 and Wenqin Wang 1,* 1 School of Agriculture and Biology, Shanghai Jiao Tong University, Shanghai 200240, China; [email protected] (Z.Z.); [email protected] (S.L.) 2 National Key Laboratory of Plant Molecular Genetics, CAS Center for Excellence in Molecular Plant Sciences, Institute of Plant Physiology & Ecology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China; [email protected] (C.J.); [email protected] (Y.Z.); [email protected] (C.L.) * Correspondence: [email protected]; Tel.: +86-21-34206942 Abstract: Improving sulfur assimilation in maize kernels is essential due to humans and animals’ inability to synthesize methionine. Serine acetyltransferase (SAT) is a critical enzyme that controls cystine biosynthesis in plants. In this study, all SAT gene members were genome-wide characterized by using a sequence homology search. The RNA-seq quantification indicates that they are highly expressed in leaves, other than root and seeds, consistent with their biological functions in sulfur assimilation. With the recently released 25 genomes of nested association mapping (NAM) founders representing the diverse maize stock, we had the opportunity to investigate the SAT genetic variation comprehensively. The abundant transposon insertions into SAT genes indicate their driving power in terms of gene structure and genome evolution. We found that the transposon insertion into exons could change SAT gene transcription, whereas there was no significant correlation between transposable element (TE) insertion into introns and their gene expression, indicating that other Citation: Zhao, Z.; Li, S.; Ji, C.; Zhou, regulatory elements such as promoters could also be involved. -

Evolution of Multigene Families by Gene Duplication: a Haploid Model
Copyright 1998 by the Genetics Society of America Evolution of Multigene Families by Gene Duplication: A Haploid Model Hidenori Tachida and Tohru Kuboyama Department of Biology, Faculty of Science, Kyushu University 33, Fukuoka 812-8581, Japan Manuscript received October 12, 1997 Accepted for publication May 13, 1998 ABSTRACT Evolution of multigene families by gene duplication and subsequent diversi®cation is analyzed assuming a haploid model without interchromosomal crossing over. Chromosomes with more different genes are assumed to have higher ®tness. Advantageous and deleterious mutations and duplication/deletion also affect the evolution, as in previous studies. In addition, negative selection on the total number of genes (copy number selection) is incorporated in the model. First, a Markov chain approximation is used to obtain formulas for the average numbers of different alleles, genes without pseudogene mutations, and pseudogenes assuming that mutation rates and duplication/deletion rates are all very small. Computer simulation shows that the approximation works well if the products of population size with mutation and duplication/deletion rates are all small compared to 1. However, as they become large, the approximation underestimates gene numbers, especially the number of pseudogenes. Based on the approximation, the following was found: (1) Gene redundancy measured by the average number of redundant genes decreases as advantageous selection becomes stronger. (2) The number of different genes can be approximately described by a linear pure-birth process and thus has a coef®cient of variation around 1. (3) The birth rate is an increasing function of population size without copy number selection, but not necessarily so otherwise. -

Ancestral Grass Karyotype Reconstruction Unravels New Mechanisms of Genome Shuffling As a Source of Plant Evolution
Downloaded from genome.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Research Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution Florent Murat,1,4 Jian-Hong Xu,2,4 Eric Tannier,3,4 Michael Abrouk,1 Nicolas Guilhot,1 Caroline Pont,1 Joachim Messing,2,5 and Je´roˆme Salse1,5 1INRA, UMR 1095, Laboratoire Ge´ne´tique, Diversite´ et Ecophysiologie des Ce´re´ales, 63100 Clermont Ferrand, France; 2The Plant Genome Initiative at Rutgers (PGIR), Waksman Institute of Microbiology, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854, USA; 3INRIA Rhoˆne-Alpes, Universite´ de Lyon 1, CNRS UMR5558, Laboratoire Biome´trie et Biologie E´volutive, 69622 Villeurbanne Cedex, France The comparison of the chromosome numbers of today’s species with common reconstructed paleo-ancestors has led to intense speculation of how chromosomes have been rearranged over time in mammals. However, similar studies in plants with respect to genome evolution as well as molecular mechanisms leading to mosaic synteny blocks have been lacking due to relevant examples of evolutionary zooms from genomic sequences. Such studies require genomes of species that belong to the same family but are diverged to fall into different subfamilies. Our most important crops belong to the family of the grasses, where a number of genomes have now been sequenced. Based on detailed paleogenomics, using inference from n = 5–12 grass ancestral karyotypes (AGKs) in terms of gene content and order, we delineated sequence intervals comprising a complete set of junction break points of orthologous regions from rice, maize, sorghum, and Brachypodium genomes, representing three different subfamilies and different polyploidization events. -

Whole-Genome Duplications and Paleopolyploidy Whole-Genome Duplications in Protozoa
Whole-genome duplications and paleopolyploidy Whole-genome duplications in protozoa •Aury et al. (2006) analyzed the unicellular eukaryote Paramecium tetraurelia • most of 40,000 genes arose through at least 3 successive whole-genome duplications • most recent duplication most likely caused an explosion of speciation events that gave rise to the P. aurelia complex (15 sibling species) • some genes have been lost, some retained • many retained (duplicated) genes do not generate functional innovations but are important because of the gene dosage effect Whole-genome duplications in yeast • genome comparison between two yeast species, Saccharomyces cerevisiae (n=16) and Kluyveromyces waltii (n=8) • each region of K. waltii corresponding to two regions of S. cerevisiae •the S. cerevisiae genome underwent a whole-genome duplication after the two yeast species diverged • in nearly every case (95%), accelerated evolution was confined to only one of the two paralogues (= one of the paralogues retained an ancestral function, the other was free to evolve more rapidly and acquired a derived function) Kellis et al. 2004, Nature 428 Whole-genome duplications in yeast Kellis et al. 2004, Nature 428 a) after divergence from K. waltii, the Saccharomyces lineage underwent a genome duplication event (2 copies of every gene and chromosome) b) duplicated genes underwent were mutated and some lost c) two copies kept for only a small minority of duplicated genes d) the conserved order of duplicated genes (nos. 3-13) across different chromosomal segments e) comparison between genomes of S. cerevisiae and K. waltii reveals the duplicated nature of the S. cerevisiae genome Duplicated nature of the S. -

Multiple Large-Scale Gene and Genome Duplications During the Evolution of Hexapods
Multiple large-scale gene and genome duplications during the evolution of hexapods Zheng Lia,1, George P. Tileyb,c,1, Sally R. Galuskaa, Chris R. Reardona, Thomas I. Kiddera, Rebecca J. Rundella,d, and Michael S. Barkera,2 aDepartment of Ecology and Evolutionary Biology, University of Arizona, Tucson, AZ 85721; bDepartment of Biology, University of Florida, Gainesville, FL 32611; cDepartment of Biology, Duke University, Durham, NC 27708; and dDepartment of Environmental and Forest Biology, State University of New York College of Environmental Science and Forestry, Syracuse, NY 13210 Edited by Michael Freeling, University of California, Berkeley, CA, and approved March 12, 2018 (received for review June 14, 2017) Polyploidy or whole genome duplication (WGD) is a major contrib- than 800,000 described hexapod species (25) are known polyploids utor to genome evolution and diversity. Although polyploidy is (17, 20). However, until recently there were limited data available recognized as an important component of plant evolution, it is to search for evidence of paleopolyploidy among the hexapods generally considered to play a relatively minor role in animal and other animal clades. Thus, the contributions of polyploidy to evolution. Ancient polyploidy is found in the ancestry of some animal evolution and the differences with plant evolution have animals, especially fishes, but there is little evidence for ancient remained unclear. WGDs in other metazoan lineages. Here we use recently published To search for evidence of WGDs among the hexapods, we transcriptomes and genomes from more than 150 species across the leveraged recently released genomic data for the insects (26). insect phylogeny to investigate whether ancient WGDs occurred Combined with additional datasets from public databases, we assembled 128 transcriptomes and 27 genomes with at least one during the evolution of Hexapoda, the most diverse clade of animals.