Multiple Large-Scale Gene and Genome Duplications During the Evolution of Hexapods
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mobile Genetic Elements in Streptococci
Curr. Issues Mol. Biol. (2019) 32: 123-166. DOI: https://dx.doi.org/10.21775/cimb.032.123 Mobile Genetic Elements in Streptococci Miao Lu#, Tao Gong#, Anqi Zhang, Boyu Tang, Jiamin Chen, Zhong Zhang, Yuqing Li*, Xuedong Zhou* State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, PR China. #Miao Lu and Tao Gong contributed equally to this work. *Address correspondence to: [email protected], [email protected] Abstract Streptococci are a group of Gram-positive bacteria belonging to the family Streptococcaceae, which are responsible of multiple diseases. Some of these species can cause invasive infection that may result in life-threatening illness. Moreover, antibiotic-resistant bacteria are considerably increasing, thus imposing a global consideration. One of the main causes of this resistance is the horizontal gene transfer (HGT), associated to gene transfer agents including transposons, integrons, plasmids and bacteriophages. These agents, which are called mobile genetic elements (MGEs), encode proteins able to mediate DNA movements. This review briefly describes MGEs in streptococci, focusing on their structure and properties related to HGT and antibiotic resistance. caister.com/cimb 123 Curr. Issues Mol. Biol. (2019) Vol. 32 Mobile Genetic Elements Lu et al Introduction Streptococci are a group of Gram-positive bacteria widely distributed across human and animals. Unlike the Staphylococcus species, streptococci are catalase negative and are subclassified into the three subspecies alpha, beta and gamma according to the partial, complete or absent hemolysis induced, respectively. The beta hemolytic streptococci species are further classified by the cell wall carbohydrate composition (Lancefield, 1933) and according to human diseases in Lancefield groups A, B, C and G. -

"Evolutionary Emergence of Genes Through Retrotransposition"
Evolutionary Emergence of Advanced article Genes Through Article Contents . Introduction Retrotransposition . Gene Alteration Following Retrotransposon Insertion . Retrotransposon Recruitment by Host Genome . Retrotransposon-mediated Gene Duplication Richard Cordaux, University of Poitiers, Poitiers, France . Conclusion Mark A Batzer, Department of Biological Sciences, Louisiana State University, Baton Rouge, doi: 10.1002/9780470015902.a0020783 Louisiana, USA Variation in the number of genes among species indicates that new genes are continuously generated over evolutionary times. Evidence is accumulating that transposable elements, including retrotransposons (which account for about 90% of all transposable elements inserted in primate genomes), are potent mediators of new gene origination. Retrotransposons have fostered genetic innovation during human and primate evolution through: (i) alteration of structure and/or expression of pre-existing genes following their insertion, (ii) recruitment (or domestication) of their coding sequence by the host genome and (iii) their ability to mediate gene duplication via ectopic recombination, sequence transduction and gene retrotransposition. Introduction genes, respectively, and de novo origination from previ- ously noncoding genomic sequence. Genome sequencing Variation in the number of genes among species indicates projects have also highlighted that new gene structures can that new genes are continuously generated over evolution- arise as a result of the activity of transposable elements ary times. Although the emergence of new genes and (TEs), which are mobile genetic units or ‘jumping genes’ functions is of central importance to the evolution of that have been bombarding the genomes of most species species, studies on the formation of genetic innovations during evolution. For example, there are over three million have only recently become possible. -

From the Human Genome Project to Genomic Medicine a Journey to Advance Human Health
From the Human Genome Project to Genomic Medicine A Journey to Advance Human Health Eric Green, M.D., Ph.D. Director, NHGRI The Origin of “Genomics”: 1987 Genomics (1987) “For the newly developing discipline of [genome] mapping/sequencing (including the analysis of the information), we have adopted the term GENOMICS… ‘The Genome Institute’ Office for Human Genome Research 1988-1989 National Center for Human Genome Research 1989-1997 National Human Genome Research Institute 1997-present NHGRI: Circa 1990-2003 Human Genome Project NHGRI Today: Characteristic Features . Relatively young (~28 years) . Relatively small (~1.7% of NIH) . Unusual historical origins (think ‘Human Genome Project’) . Emphasis on ‘Team Science’ (think managed ‘consortia’) . Rapidly disseminating footprint (think ‘genomics’) . Novel societal/bioethics research component (think ‘ELSI’) . Over-achievers for trans-NIH initiatives (think ‘Common Fund’) . Vibrant (and large) Intramural Research Program A Quarter Century of Genomics Human Genome Sequenced for First Time by the Human Genome Project Genomic Medicine An emerging medical discipline that involves using genomic information about an individual as part of their clinical care (e.g., for diagnostic or therapeutic decision- making) and the other implications of that clinical use The Path to Genomic Medicine ? Human Realization of Genome Genomic Project Medicine Nature Nature Base Pairs to Bedside 2003 Heli201x to 1Health A Quarter Century of Genomics Human Genome Sequenced for First Time by the Human Genome Project -
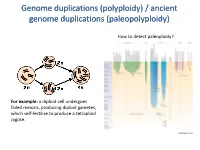
Polyploidy) / Ancient Genome Duplications (Paleopolyploidy
Genome duplications (polyploidy) / ancient genome duplications (paleopolyploidy) How to detect paleoploidy? For example: a diploid cell undergoes failed meiosis, producing diploid gametes, which self-fertilize to produce a tetraploid zygote. Timing of duplication by trees (phylogenetic timing) Phylogenetic timing of duplicates b Paramecium genome duplications Comparison of two scaffolds originating from a common ancestor at the recent WGD Saccharomyces cerevisiae Just before genome duplication Just after genome duplication More time after genome duplication Unaligned view (removing gaps just like in cerev has occurred) Saccharomyces cerevisiae Problem reciprocal gene loss (extreme case); how to solve? Problem reciprocal gene loss (extreme case); how to solve? Just before genome duplication Outgroup! Just after genome duplication Outgroup Just after genome duplication Outgroup More time after genome duplication Outgroup Problem (extreme case); how to solve? Outgroup Outgroup Outgroup Outgroup Outgroup Using other genomes Wong et al. 2002 PNAS Centromeres Vertebrate genome duplication Nature. 2011 Apr 10. [Epub ahead of print] Ancestral polyploidy in seed plants and angiosperms. Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, Depamphilis CW. Flowering plants Flowering MOSS Vertebrates Teleosts S. serevisiae and close relatives Paramecium Reconstructed map of genome duplications allows unprecedented mapping -

The Diversity of Plant Sex Chromosomes Highlighted Through Advances in Genome Sequencing
G C A T T A C G G C A T genes Review The Diversity of Plant Sex Chromosomes Highlighted through Advances in Genome Sequencing Sarah Carey 1,2 , Qingyi Yu 3,* and Alex Harkess 1,2,* 1 Department of Crop, Soil, and Environmental Sciences, Auburn University, Auburn, AL 36849, USA; [email protected] 2 HudsonAlpha Institute for Biotechnology, Huntsville, AL 35806, USA 3 Texas A&M AgriLife Research, Texas A&M University System, Dallas, TX 75252, USA * Correspondence: [email protected] (Q.Y.); [email protected] (A.H.) Abstract: For centuries, scientists have been intrigued by the origin of dioecy in plants, characterizing sex-specific development, uncovering cytological differences between the sexes, and developing theoretical models. Through the invention and continued improvements in genomic technologies, we have truly begun to unlock the genetic basis of dioecy in many species. Here we broadly review the advances in research on dioecy and sex chromosomes. We start by first discussing the early works that built the foundation for current studies and the advances in genome sequencing that have facilitated more-recent findings. We next discuss the analyses of sex chromosomes and sex-determination genes uncovered by genome sequencing. We synthesize these results to find some patterns are emerging, such as the role of duplications, the involvement of hormones in sex-determination, and support for the two-locus model for the origin of dioecy. Though across systems, there are also many novel insights into how sex chromosomes evolve, including different sex-determining genes and routes to suppressed recombination. We propose the future of research in plant sex chromosomes should involve interdisciplinary approaches, combining cutting-edge technologies with the classics Citation: Carey, S.; Yu, Q.; to unravel the patterns that can be found across the hundreds of independent origins. -

A 'David and Goliath'
Current Genomics, 2002, 3, 563-576 563 Nucleolar Dominance: A ‘David and Goliath’ Chromatin Imprinting Process W. Viegas1,*, N. Neves1,2, M. Silva1, A. Caperta1,2, and L. Morais-Cecílio1 1Centro de Botânica Aplicada à Agricultura, Secção de Genética/DBEB, Instituto Superior de Agronomia, 1349-017 Lisboa, 2Departamento de Ciências Biológicas e Naturais, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande 376, 1749-042 Lisboa, Portugal Abstract: Nucleolar dominance is an enigma. The puzzle of differential amphiplasty has remained unresolved since it was first recognised and described in Crepis hybrids by Navashin in 1934. Here we review the body of knowledge that has grown out of the many models that have tried to find the genetic basis for differential rRNA gene expression in hybrids, and present a new interpretation. We propose and discuss a chromatin imprinting model which re-interprets differential amphiplasty in terms of two genomes of differing size occupying a common space within the nucleus, and with heterochromatin as a key player in the scenario. Difference in size between two parental genomes induces an inherited epigenetic mark in the hybrid that allows patterns of chromatin organization to have positional effects on the neighbouring domains. This chromatin imprinting model can be also used to explain complex genomic interactions which transcend nucleolar dominance and which can account for the overall characteristics of hybrids. Gene expression in hybrids, relative to parentage, is seen as being based on the nuclear location of the sequences concerned within their genomic environment, and where the presence of particular repetitive DNA sequences are ‘sensed’, and render silent the adjacent information. -

Gene Loss and Adaptation in Saccharomyces Genomes
Genetics: Published Articles Ahead of Print, published on December 1, 2005 as 10.1534/genetics.105.048900 After the duplication: gene loss and adaptation in Saccharomyces genomes Paul F. Cliften*,1, Robert S. Fulton§, Richard K. Wilson*, §, and Mark Johnston* *Department of Genetics and §Genome Sequencing Center, Washington University School of Medicine, 660 South Euclid Ave., St. Louis, MO, 63110; 1Current address: Department of Biology, Utah State University, 5305 Old Main Hill, Logan, UT, 84322 Running head: Saccharomyces genomic duplications Key words: genomic duplication, comparative sequence analysis, Saccharomyces phylogeny, yeast Corresponding author: Mark Johnston Department of Genetics Campus Box 8232 Washington University Medical School 4566 Scott Ave. St. Louis, MO 63110 TEL: 314-362-2735 FAX: 314-362-2157 [email protected] ABSTRACT The ancient duplication of the Saccharomyces cerevisiae genome and subsequent massive loss of duplicated genes is apparent when it is compared to the genomes of related species that diverged before the duplication event. To learn more about the evolutionary effects of the duplication event, we compared the S. cerevisiae genome to other Saccharomyces genomes. We demonstrate that the whole genome duplication occurred before S. castellii diverged from S. cerevisiae. In addition to more accurately dating the duplication event, this finding allowed us to study the effects of the duplication on two separate lineages. Analyses of the duplication regions of the genomes indicate that most of the duplicated genes (approximately 85%) were lost before the speciation. Only a small amount of paralogous gene loss (4-6%) occurred after speciation. On the other hand, S. castellii appears to have lost several hundred genes that were not retained as duplicated paralogs. -

2R Or Not 2R Is Not the Question Anymore
CORRESPONDENCE LINK TO ORIGINAL ARTICLE LINK TO INITIAL CORRESPONDENCE origin of evolutionary novelties are highly interesting questions, but they remain 2R or not 2R is not the largely unsolved11. However, evidence for the two rounds of genome duplication dur- question anymore ing chordate evolution is very strong, and it would seem safe to say that the debate Yves Van de Peer, Steven Maere and Axel Meyer over 2R is settled and is no longer an open question12. As we point out in our Opinion article1, it is only the evolutionary effects of In his comments on our Opinion article in passing in our recent review on WGDs events such as WGDs on evolution that are (The evolutionary significance of ancient and their significance for evolution in Nature debated1,11, and no longer whether or not genome duplications. Nature Rev. Genet. Reviews Genetics1. two rounds of WGD occurred during the 10, 725–732 (2009))1 Amir Ali Abbasi Abbasi2, however, questions the support evolution of chordates. (Piecemeal or big bangs: correlating the for the 2R hypothesis, claiming that it still Yves Van de Peer and Steven Maere are at the vertebrate evolution with proposed models remains debated today. He points out cor- Department of Plant Systems Biology, VIB (Flanders of gene expansion events. Nature Rev. Genet. rectly that evidence for the 2R hypothesis Institute of Biotechnology), B-9052 Ghent, Belgium. 6 Jan 2010 (doi:10.1038/nrg2600-c1))2 was based initially on data from only a Axel Meyer is at the Department of Biology, University argues that it is not justified to speculate small number of genes and vertebrate and of Konstanz, D-78457 Konstanz, Germany. -

Iv Gene Duplication and the Evolution of Silenced
Gene Duplication and the Evolution of Silenced Chromatin in Yeasts by Meleah A. Hickman University Program in Genetics and Genomics Duke University Date:_______________________ Approved: ___________________________ Laura Rusche, Advisor ___________________________ Paul Magwene, Chair ___________________________ Fred Dietrich ___________________________ Joe Heitman ___________________________ Beth Sullivan Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2010 i v ABSTRACT Gene Duplication and the Evolution of Silenced Chromatin in Yeasts by Meleah A. Hickman University Program in Genetics and Genomics Duke University Date:_______________________ Approved: ___________________________ Laura Rusche, Advisor ___________________________ Paul Magwene, Chair ___________________________ Fred Dietrich ___________________________ Joe Heitman ___________________________ Beth Sullivan An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2010 Copyright by Meleah A. Hickman 2010 Abstract In Saccharomyces cerevisiae, proper maintenance of haploid cell identity requires the SIR complex to mediate the silenced chromatin found at the cryptic mating-type loci, HML and HMR. This complex consists of Sir2, a histone deacetylase, and the histone binding proteins Sir3 and Sir4. Interestingly, both Sir2 and Sir3 have paralogs from a genome duplication that occurred after the divergence of Saccharomyces and Kluyveromyces species, approximately 100 million years ago. The histone deacetylase HST1 is the paralog of SIR2 and works with the promoter-specific SUM1 complex to repress sporulation and alpha-specific genes. ORC1 is the paralog of SIR3 and is an essential subunit of the Origin Recognition Complex and also recruits SIR proteins to the HM loci. -

A Study of Visitors to Genome: the Secret of How Life Works
A STUDY OF VISITORS TO GENOME: THE SECRET OF HOW LIFE WORKS March 2004 Office of Policy and Analysis Washington, DC 20560-0405 Office of Policy and Analysis Study Team Ioana Munteanu Andrew Pekarik Whitney Watriss FOREWORD This study of Genome: The Secret of How Life Works focuses on visitors’ perceptions of the exhibition. It draws attention to interactive aspects of the exhibition as well as to underlying conceptual dimensions: basic genome science and applied genome science. To produce information on the success of the exhibition, it captures responses to key factors in the exhibit, such as visitor satisfaction with respect to interactive activities and visitor satisfaction concerning physical attributes and explores the relationships among these factors through correlational analysis. The study was designed and administered by Ioana Munteanu, who analyzed the data and wrote this report; several colleagues from the Office of Policy and Analysis offered helpful advice on various parts of the study. Carole M. P. Neves Director Office of Policy and Analysis PART I. BACKGROUND Fifty years ago, Nobel Prize winners Francis Crick and James Watson first visualized the three-dimensional molecular structure of deoxyribonucleic acid’s (DNA) double helix. Since they accomplished that extraordinary feat, scientists have made remarkable progress in the field of modern genomics, including sequencing the human genome.1 The traveling exhibition, Genome: The Secret of How Life Works, sponsored by Pfizer and produced by Clear Channel Entertainment-Exhibition, opened for display in a 5,000- square foot gallery in the Smithsonian Institution’s Arts and Industries Building (A&I Building) on June 6, 2003. -

The Economic Impact and Functional Applications of Human Genetics and Genomics
The Economic Impact and Functional Applications of Human Genetics and Genomics Commissioned by the American Society of Human Genetics Produced by TEConomy Partners, LLC. Report Authors: Simon Tripp and Martin Grueber May 2021 TEConomy Partners, LLC (TEConomy) endeavors at all times to produce work of the highest quality, consistent with our contract commitments. However, because of the research and/or experimental nature of this work, the client undertakes the sole responsibility for the consequence of any use or misuse of, or inability to use, any information or result obtained from TEConomy, and TEConomy, its partners, or employees have no legal liability for the accuracy, adequacy, or efficacy thereof. Acknowledgements ASHG and the project authors wish to thank the following organizations for their generous support of this study. Invitae Corporation, San Francisco, CA Regeneron Pharmaceuticals, Inc., Tarrytown, NY The project authors express their sincere appreciation to the following indi- viduals who provided their advice and input to this project. ASHG Government and Public Advocacy Committee Lynn B. Jorde, PhD ASHG Government and Public Advocacy Committee (GPAC) Chair, President (2011) Professor and Chair of Human Genetics George and Dolores Eccles Institute of Human Genetics University of Utah School of Medicine Katrina Goddard, PhD ASHG GPAC Incoming Chair, Board of Directors (2018-2020) Distinguished Investigator, Associate Director, Science Programs Kaiser Permanente Northwest Melinda Aldrich, PhD, MPH Associate Professor, Department of Medicine, Division of Genetic Medicine Vanderbilt University Medical Center Wendy Chung, MD, PhD Professor of Pediatrics in Medicine and Director, Clinical Cancer Genetics Columbia University Mira Irons, MD Chief Health and Science Officer American Medical Association Peng Jin, PhD Professor and Chair, Department of Human Genetics Emory University Allison McCague, PhD Science Policy Analyst, Policy and Program Analysis Branch National Human Genome Research Institute Rebecca Meyer-Schuman, MS Human Genetics Ph.D. -

Genome Organization/ Human
Genome Organization/ Secondary article Human Article Contents . Introduction David H Kass, Eastern Michigan University, Ypsilanti, Michigan, USA . Sequence Complexity Mark A Batzer, Louisiana State University Health Sciences Center, New Orleans, Louisiana, USA . Single-copy Sequences . Repetitive Sequences . The human nuclear genome is a highly complex arrangement of two sets of 23 Macrosatellites, Minisatellites and Microsatellites . chromosomes, or DNA molecules. There are various types of DNA sequences and Gene Families . chromosomal arrangements, including single-copy protein-encoding genes, repetitive Gene Superfamilies . sequences and spacer DNA. Transposable Elements . Pseudogenes . Mitochondrial Genome Introduction . Genome Evolution . Acknowledgements The human nuclear genome contains 3000 million base pairs (bp) of DNA, of which only an estimated 3% possess protein-encoding sequences. As shown in Figure 1, the DNA sequences of the eukaryotic genome can be classified sequences such as the ribosomal RNA genes. Repetitive into several types, including single-copy protein-encoding sequences with no known function include the various genes, DNA that is present in more than one copy highly repeated satellite families, and the dispersed, (repetitive sequences) and intergenic (spacer) DNA. The moderately repeated transposable element families. The most complex of these are the repetitive sequences, some of remainder of the genome consists of spacer DNA, which is which are functional and some of which are without simply a broad category of undefined DNA sequences. function. Functional repetitive sequences are classified into The human nuclear genome consists of 23 pairs of dispersed and/or tandemly repeated gene families that chromosomes, or 46 DNA molecules, of differing sizes either encode proteins (and may include noncoding (Table 1).