Ancestral Grass Karyotype Reconstruction Unravels New Mechanisms of Genome Shuffling As a Source of Plant Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma
cancers Article Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma 1, 1, 1 1 1 Lan Wang y, Anudeep Yekula y, Koushik Muralidharan , Julia L. Small , Zachary S. Rosh , Keiko M. Kang 1,2, Bob S. Carter 1,* and Leonora Balaj 1,* 1 Department of Neurosurgery, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02115, USA; [email protected] (L.W.); [email protected] (A.Y.); [email protected] (K.M.); [email protected] (J.L.S.); [email protected] (Z.S.R.); [email protected] (K.M.K.) 2 School of Medicine, University of California San Diego, San Diego, CA 92092, USA * Correspondence: [email protected] (B.S.C.); [email protected] (L.B.) These authors contributed equally. y Received: 11 March 2020; Accepted: 7 May 2020; Published: 13 May 2020 Abstract: Sequencing studies have provided novel insights into the heterogeneous molecular landscape of glioblastoma (GBM), unveiling a subset of patients with gene fusions. Tissue biopsy is highly invasive, limited by sampling frequency and incompletely representative of intra-tumor heterogeneity. Extracellular vesicle-based liquid biopsy provides a minimally invasive alternative to diagnose and monitor tumor-specific molecular aberrations in patient biofluids. Here, we used targeted RNA sequencing to screen GBM tissue and the matched plasma of patients (n = 9) for RNA fusion transcripts. We identified two novel fusion transcripts in GBM tissue and five novel fusions in the matched plasma of GBM patients. The fusion transcripts FGFR3-TACC3 and VTI1A-TCF7L2 were detected in both tissue and matched plasma. -
Polyploidy) / Ancient Genome Duplications (Paleopolyploidy
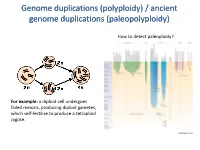
Genome duplications (polyploidy) / ancient genome duplications (paleopolyploidy) How to detect paleoploidy? For example: a diploid cell undergoes failed meiosis, producing diploid gametes, which self-fertilize to produce a tetraploid zygote. Timing of duplication by trees (phylogenetic timing) Phylogenetic timing of duplicates b Paramecium genome duplications Comparison of two scaffolds originating from a common ancestor at the recent WGD Saccharomyces cerevisiae Just before genome duplication Just after genome duplication More time after genome duplication Unaligned view (removing gaps just like in cerev has occurred) Saccharomyces cerevisiae Problem reciprocal gene loss (extreme case); how to solve? Problem reciprocal gene loss (extreme case); how to solve? Just before genome duplication Outgroup! Just after genome duplication Outgroup Just after genome duplication Outgroup More time after genome duplication Outgroup Problem (extreme case); how to solve? Outgroup Outgroup Outgroup Outgroup Outgroup Using other genomes Wong et al. 2002 PNAS Centromeres Vertebrate genome duplication Nature. 2011 Apr 10. [Epub ahead of print] Ancestral polyploidy in seed plants and angiosperms. Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, Soltis DE, Clifton SW, Schlarbaum SE, Schuster SC, Ma H, Leebens-Mack J, Depamphilis CW. Flowering plants Flowering MOSS Vertebrates Teleosts S. serevisiae and close relatives Paramecium Reconstructed map of genome duplications allows unprecedented mapping -

A 'David and Goliath'
Current Genomics, 2002, 3, 563-576 563 Nucleolar Dominance: A ‘David and Goliath’ Chromatin Imprinting Process W. Viegas1,*, N. Neves1,2, M. Silva1, A. Caperta1,2, and L. Morais-Cecílio1 1Centro de Botânica Aplicada à Agricultura, Secção de Genética/DBEB, Instituto Superior de Agronomia, 1349-017 Lisboa, 2Departamento de Ciências Biológicas e Naturais, Universidade Lusófona de Humanidades e Tecnologias, Campo Grande 376, 1749-042 Lisboa, Portugal Abstract: Nucleolar dominance is an enigma. The puzzle of differential amphiplasty has remained unresolved since it was first recognised and described in Crepis hybrids by Navashin in 1934. Here we review the body of knowledge that has grown out of the many models that have tried to find the genetic basis for differential rRNA gene expression in hybrids, and present a new interpretation. We propose and discuss a chromatin imprinting model which re-interprets differential amphiplasty in terms of two genomes of differing size occupying a common space within the nucleus, and with heterochromatin as a key player in the scenario. Difference in size between two parental genomes induces an inherited epigenetic mark in the hybrid that allows patterns of chromatin organization to have positional effects on the neighbouring domains. This chromatin imprinting model can be also used to explain complex genomic interactions which transcend nucleolar dominance and which can account for the overall characteristics of hybrids. Gene expression in hybrids, relative to parentage, is seen as being based on the nuclear location of the sequences concerned within their genomic environment, and where the presence of particular repetitive DNA sequences are ‘sensed’, and render silent the adjacent information. -

Extending the Phenotypic Spectrum of Sengers Syndrome: Congenital Lactic Acidosis with Synthetic Liver Dysfunction
Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pathology Faculty Publications Pathology 4-13-2018 Extending the phenotypic spectrum of Sengers syndrome: Congenital lactic acidosis with synthetic liver dysfunction. David B Beck Kristina Cusmano-Ozog George Washington University Nickie Andescavage George Washington University Eyby Leon George Washington University Follow this and additional works at: https://hsrc.himmelfarb.gwu.edu/smhs_path_facpubs Part of the Medical Pathology Commons, Pathology Commons, and the Translational Medical Research Commons APA Citation Beck, D., Cusmano-Ozog, K., Andescavage, N., & Leon, E. (2018). Extending the phenotypic spectrum of Sengers syndrome: Congenital lactic acidosis with synthetic liver dysfunction.. Translational Science of Rare Diseases, 3 (1). http://dx.doi.org/10.3233/ TRD-180020 This Journal Article is brought to you for free and open access by the Pathology at Health Sciences Research Commons. It has been accepted for inclusion in Pathology Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. Translational Science of Rare Diseases 3 (2018) 45–48 45 DOI 10.3233/TRD-180020 IOS Press Original Research Extending the phenotypic spectrum of Sengers syndrome: Congenital lactic acidosis with synthetic liver dysfunction David B. Becka, Kristina Cusmano-Ozogb, Nickie Andescavagec and Eyby Leonb,∗ aNational Human Genome Research Institute, National Institute of Health, Bethesda, MD, USA bChildren’s National Health System, Rare Disease Institute, Genetics and Metabolism, Washington, DC, USA cChildren’s National Health System, Pediatrics, Neonatology, Washington, DC, USA Abstract. Sengers syndrome is a rare autosomal recessive mitochondrial disease characterized by lactic acidosis, hypertrophic cardiomyopathy and bilateral cataracts. -

Gene Loss and Adaptation in Saccharomyces Genomes
Genetics: Published Articles Ahead of Print, published on December 1, 2005 as 10.1534/genetics.105.048900 After the duplication: gene loss and adaptation in Saccharomyces genomes Paul F. Cliften*,1, Robert S. Fulton§, Richard K. Wilson*, §, and Mark Johnston* *Department of Genetics and §Genome Sequencing Center, Washington University School of Medicine, 660 South Euclid Ave., St. Louis, MO, 63110; 1Current address: Department of Biology, Utah State University, 5305 Old Main Hill, Logan, UT, 84322 Running head: Saccharomyces genomic duplications Key words: genomic duplication, comparative sequence analysis, Saccharomyces phylogeny, yeast Corresponding author: Mark Johnston Department of Genetics Campus Box 8232 Washington University Medical School 4566 Scott Ave. St. Louis, MO 63110 TEL: 314-362-2735 FAX: 314-362-2157 [email protected] ABSTRACT The ancient duplication of the Saccharomyces cerevisiae genome and subsequent massive loss of duplicated genes is apparent when it is compared to the genomes of related species that diverged before the duplication event. To learn more about the evolutionary effects of the duplication event, we compared the S. cerevisiae genome to other Saccharomyces genomes. We demonstrate that the whole genome duplication occurred before S. castellii diverged from S. cerevisiae. In addition to more accurately dating the duplication event, this finding allowed us to study the effects of the duplication on two separate lineages. Analyses of the duplication regions of the genomes indicate that most of the duplicated genes (approximately 85%) were lost before the speciation. Only a small amount of paralogous gene loss (4-6%) occurred after speciation. On the other hand, S. castellii appears to have lost several hundred genes that were not retained as duplicated paralogs. -

Two Novel Mutations in the AGK Gene: Two Case Reports with Sengers
Techno e lo Kor et al., Gene Technol 2016, 5:1 n g e y G Gene Technology DOI: 10.4172/2329-6682.1000140 ISSN: 2329-6682 Case Report Open Access Two Novel Mutations in the AGK Gene: Two Case Reports with Sengers Syndrome Deniz Kor1*, Berna Seker Yılmaz1, Ozden Ozgur Horoz2, Gulay Ceylaner3, Selcuk Sızmaz4, Fadli Demir2 and Neslihan Onenli Mungan1 1Department of Pediatric Metabolism and Nutrition, Faculty of Medicine, Cukurova University, Adana, Turkey 2Department of Pediatrics, Faculty of Medicine, Cukurova University, Adana, Turkey 3Intergen Genetic Laboratory, Ankara, Turkey 4Department of Ophthalmology, Cukurova University Faculty of Medicine, Adana, Turkey Abstract Mutations in the AGK gene are known to cause Sengers Syndrome, a rare recessive disorder characterized by congenital cataracts, hypertrophic cardiomyopathy, skeletal myopathy, exercise intolerance and lactic acidosis with normal mental development. Since the first report in 1975 by Sengers et al. about 50 individuals have been described as having this syndrome. Here we report two novel mutations in the AGK gene in two patients with neonatal Sengers syndrome. Keywords: AGK gene; Sengers syndrome; Cardiomyopathy; acylcarnitine levels, plasma amino acids and urine organic acids) Myopathy; Cataract were within normal limits except elevated levels of creatine kinases and lactate. Echocardiographic evaluation documented hypertrophic Introduction cardiomyopathy with an ejection fraction of 30%. Electromyography Sengers Syndrome (OMIM 212350), also known as cardiomyopathic and cerebral MRI showed no abnormality. A quadriceps muscle biopsy mitochondrial DNA depletion syndrome-10 (MTDPS10) is a rare revealed fatty infiltrations in the muscle fibers and myopathic changes. autosomal-recessive disorder characterized by congenital cataracts, Based on these observations a clinical diagnosis of Sengers Syndrome hypertrophic cardiomyopathy, skeletal myopathy, exercise intolerance was made. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Iv Gene Duplication and the Evolution of Silenced
Gene Duplication and the Evolution of Silenced Chromatin in Yeasts by Meleah A. Hickman University Program in Genetics and Genomics Duke University Date:_______________________ Approved: ___________________________ Laura Rusche, Advisor ___________________________ Paul Magwene, Chair ___________________________ Fred Dietrich ___________________________ Joe Heitman ___________________________ Beth Sullivan Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2010 i v ABSTRACT Gene Duplication and the Evolution of Silenced Chromatin in Yeasts by Meleah A. Hickman University Program in Genetics and Genomics Duke University Date:_______________________ Approved: ___________________________ Laura Rusche, Advisor ___________________________ Paul Magwene, Chair ___________________________ Fred Dietrich ___________________________ Joe Heitman ___________________________ Beth Sullivan An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the University Program in Genetics and Genomics in the Graduate School of Duke University 2010 Copyright by Meleah A. Hickman 2010 Abstract In Saccharomyces cerevisiae, proper maintenance of haploid cell identity requires the SIR complex to mediate the silenced chromatin found at the cryptic mating-type loci, HML and HMR. This complex consists of Sir2, a histone deacetylase, and the histone binding proteins Sir3 and Sir4. Interestingly, both Sir2 and Sir3 have paralogs from a genome duplication that occurred after the divergence of Saccharomyces and Kluyveromyces species, approximately 100 million years ago. The histone deacetylase HST1 is the paralog of SIR2 and works with the promoter-specific SUM1 complex to repress sporulation and alpha-specific genes. ORC1 is the paralog of SIR3 and is an essential subunit of the Origin Recognition Complex and also recruits SIR proteins to the HM loci. -

Identification of Kinase Fusion Oncogenes in Post-Chernobyl Radiation-Induced Thyroid Cancers
Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers Julio C. Ricarte-Filho, … , Christopher E. Mason, James A. Fagin J Clin Invest. 2013;123(11):4935-4944. https://doi.org/10.1172/JCI69766. Research Article Exposure to ionizing radiation during childhood markedly increases the risk of developing papillary thyroid cancer. We examined tissues from 26 Ukrainian patients with thyroid cancer who were younger than 10 years of age and living in contaminated areas during the time of the Chernobyl nuclear reactor accident. We identified nonoverlapping somatic driver mutations in all 26 cases through candidate gene assays and next-generation RNA sequencing. We found that 22 tumors harbored fusion oncogenes that arose primarily through intrachromosomal rearrangements. Altogether, 23 of the oncogenic drivers identified in this cohort aberrantly activate MAPK signaling, including the 2 somatic rearrangements resulting in fusion of transcription factor ETS variant 6 (ETV6) with neurotrophic tyrosine kinase receptor, type 3 (NTRK3) and fusion of acylglycerol kinase (AGK) with BRAF. Two other tumors harbored distinct fusions leading to overexpression of the nuclear receptor PPARγ. Fusion oncogenes were less prevalent in tumors from a cohort of children with pediatric thyroid cancers that had not been exposed to radiation but were from the same geographical regions. Radiation-induced thyroid cancers provide a paradigm of tumorigenesis driven by fusion oncogenes that activate MAPK signaling or, less frequently, a PPARγ-driven transcriptional program. Find the latest version: https://jci.me/69766/pdf Related Commentary, page 4566 Research article Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers Julio C. -

Whole-Genome Duplications and Paleopolyploidy Whole-Genome Duplications
Whole-genome duplications and paleopolyploidy Whole-genome duplications AUTOPOLYPLOIDY ALLOPOLYPLOIDY Hegarty and HiscockHegarty 2008, 18 Current Biology Examples of allopolyploid speciation Hegarty and Hiscock 2008, Current Biology 18 Whole-genome duplications of different age time neopolyploidy mesopolyploidy paleopolyploidy time Whole-genome duplications in protozoa •Aury et al. (2006) analyzed the unicellular eukaryote Paramecium tetraurelia • most of 40,000 genes arose through at least 3 successive whole-genome duplications (WGDs) • most recent duplication most likely caused an explosion of speciation events that gave rise to the P. aurelia complex (15 sibling species) • some genes have been lost, some retained • many retained (duplicated) genes do not generate functional innovations but are important because of the gene dosage effect Whole-genome duplications in yeast • genome comparison between two yeast species, Saccharomyces cerevisiae (n=16) and Kluyveromyces waltii (n=8) • each region of K. waltii corresponding to two regions of S. cerevisiae •the S. cerevisiae genome underwent a WGD after the two yeast species diverged • in nearly every case (95%), accelerated evolution was confined to only one of the two paralogues (= one of the paralogues retained an ancestral function, the other was free to evolve more rapidly and acquired a derived function) Kellis et al. 2004, Nature 428 Whole-genome duplications in yeast Kellis et al. 2004, Nature 428 a) after divergence from K. waltii, the Saccharomyces lineage underwent a genome duplication event (2 copies of every gene and chromosome) b) duplicated genes were mutated and some lost c) two copies kept for only a small minority of duplicated genes d) the conserved order of duplicated genes (nos. -

Transposable Elements in Polyploid Evolution Final.Pdf
Transposable elements and polyploid evolution in animals Fernando Rodriguez and Irina R. Arkhipova Address Josephine Bay Paul Center for Comparative Molecular Biology and Evolution, Marine Biological Laboratory, Woods Hole, MA 02543, USA Corresponding author: Arkhipova, Irina R. ([email protected]) Polyploidy in animals is much less common than in plants, where it is thought to be pervasive in all higher plant lineages. Recent studies have highlighted the impact of polyploidization and the associated process of diploidy restoration on the evolution and speciation of selected taxonomic groups in the animal kingdom: from vertebrates represented by salmonid fishes and African clawed frogs to invertebrates represented by parasitic root-knot nematodes and bdelloid rotifers. In this review, we focus on the unique and diverse roles that transposable elements may play in these processes, from marking and diversifying subgenome-specific chromosome sets prior to hybridization, to influencing genome restructuring during rediploidization, to affecting subgenome-specific regulatory evolution, and occasionally providing opportunities for domestication and gene amplification to restore and improve functionality. There is still much to be learned from the future comparative genomic studies of chromosome-sized and haplotype-aware assemblies, and from post-genomic studies elucidating genetic and epigenetic regulatory phenomena across short and long evolutionary distances in the metazoan tree of life. Glossary Polyploidy: having more than the usual diploid set of homologous chromosomes Allopolyploidy: a form of polyploidy that results from interspecific hybridization Autopolyploidy: a form of polyploidy that results from chromosome redoubling in the same species Paleopolyploidy: ancient polyploidy Neopolyploidy: recent polyploidy Homeologs: pairs of genes originated by speciation and brought back together by allopolyploidization Ohnologs: same as homeologs, named after S. -

The Chromosome-Based Lavender Genome Provides New Insights Into
Li et al. Horticulture Research (2021) 8:53 Horticulture Research https://doi.org/10.1038/s41438-021-00490-6 www.nature.com/hortres ARTICLE Open Access The chromosome-based lavender genome provides new insights into Lamiaceae evolution and terpenoid biosynthesis Jingrui Li1,2, Yiming Wang 3,YanmeiDong1,2, Wenying Zhang1,2,DiWang1, Hongtong Bai1,KuiLi3,HuiLi1 and Lei Shi1 Abstract The aromatic shrub Lavandula angustifolia produces various volatile terpenoids that serve as resources for essential oils and function in plant-insect communication. To better understand the genetic basis of the terpenoid diversity in lavender, we present a high-quality reference genome for the Chinese lavender cultivar ‘Jingxun 2’ using PacBio and Hi-C technologies to anchor the 894.50 Mb genome assembly into 27 pseudochromosomes. In addition to the γ triplication event, lavender underwent two rounds of whole-genome duplication (WGD) during the Eocene–Oligocene (29.6 MYA) and Miocene–Pliocene (6.9 MYA) transitions. As a result of tandem duplications and lineage-specific WGDs, gene families related to terpenoid biosynthesis in lavender are substantially expanded compared to those of five other species in Lamiaceae. Many terpenoid biosynthesis transcripts are abundant in glandular trichomes. We further integrated the contents of ecologically functional terpenoids and coexpressed terpenoid biosynthetic genes to construct terpenoid-gene networks. Typical gene clusters, including TPS-TPS, TPS- CYP450, and TPS-BAHD, linked with compounds that primarily function as attractants or repellents, were identified by fl 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; their similar patterns of change during ower development or in response to methyl jasmonate. Comprehensive analysis of the genetic basis of the production of volatiles in lavender could serve as a foundation for future research into lavender evolution, phytochemistry, and ecology.