Statistical Methods for Predicting Genetic Regulation by Nisar Ahmed
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Gene Expression Profile in Different Age Groups and Its Association With
cells Article Gene Expression Profile in Different Age Groups and Its Association with Cognitive Function in Healthy Malay Adults in Malaysia Nur Fathiah Abdul Sani 1 , Ahmad Imran Zaydi Amir Hamzah 1, Zulzikry Hafiz Abu Bakar 1 , Yasmin Anum Mohd Yusof 2, Suzana Makpol 1 , Wan Zurinah Wan Ngah 1 and Hanafi Ahmad Damanhuri 1,* 1 Department of Biochemistry, Faculty of Medicine, Universiti Kebangsaan Malaysia Medical Center, Jalan Yaacob Latif, Cheras, Kuala Lumpur 56000, Malaysia; [email protected] (N.F.A.S.); [email protected] (A.I.Z.A.H.); zulzikryhafi[email protected] (Z.H.A.B.); [email protected] (S.M.); [email protected] (W.Z.W.N.) 2 Faculty of Medicine and Defence Health, National Defence University of Malaysia, Kem Sungai Besi, Kuala Lumpur 57000, Malaysia; [email protected] * Correspondence: hanafi[email protected] Abstract: The mechanism of cognitive aging at the molecular level is complex and not well under- stood. Growing evidence suggests that cognitive differences might also be caused by ethnicity. Thus, this study aims to determine the gene expression changes associated with age-related cognitive decline among Malay adults in Malaysia. A cross-sectional study was conducted on 160 healthy Malay subjects, aged between 28 and 79, and recruited around Selangor and Klang Valley, Malaysia. Citation: Abdul Sani, N.F.; Amir Gene expression analysis was performed using a HumanHT-12v4.0 Expression BeadChip microarray Hamzah, A.I.Z.; Abu Bakar, Z.H.; kit. The top 20 differentially expressed genes at p < 0.05 and fold change (FC) = 1.2 showed that Mohd Yusof, Y.A.; Makpol, S.; Wan PAFAH1B3, HIST1H1E, KCNA3, TM7SF2, RGS1, and TGFBRAP1 were regulated with increased Ngah, W.Z.; Damanhuri, H.A. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -
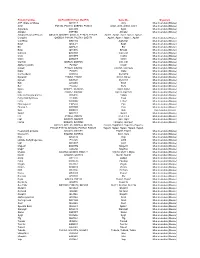
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures. -

Revealing Transcription Factor and Histone Modification Co-Localization and Dynamics Across Cell Lines by Integrating Chip-Seq A
Zhang et al. BMC Genomics 2018, 19(Suppl 10):914 https://doi.org/10.1186/s12864-018-5278-5 RESEARCH Open Access Revealing transcription factor and histone modification co-localization and dynamics across cell lines by integrating ChIP-seq and RNA-seq data Lirong Zhang1*, Gaogao Xue1, Junjie Liu1, Qianzhong Li1* and Yong Wang2,3,4* From 29th International Conference on Genome Informatics Yunnan, China. 3-5 December 2018 Abstract Background: Interactions among transcription factors (TFs) and histone modifications (HMs) play an important role in the precise regulation of gene expression. The context specificity of those interactions and further its dynamics in normal and disease remains largely unknown. Recent development in genomics technology enables transcription profiling by RNA-seq and protein’s binding profiling by ChIP-seq. Integrative analysis of the two types of data allows us to investigate TFs and HMs interactions both from the genome co-localization and downstream target gene expression. Results: We propose a integrative pipeline to explore the co-localization of 55 TFs and 11 HMs and its dynamics in human GM12878 and K562 by matched ChIP-seq and RNA-seq data from ENCODE. We classify TFs and HMs into three types based on their binding enrichment around transcription start site (TSS). Then a set of statistical indexes are proposed to characterize the TF-TF and TF-HM co-localizations. We found that Rad21, SMC3, and CTCF co-localized across five cell lines. High resolution Hi-C data in GM12878 shows that they associate most of the Hi-C peak loci with a specific CTCF-motif “anchor” and supports that CTCF, SMC3, and RAD2 co-localization serves important role in 3D chromatin structure. -

Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Integration of 198 ChIP-seq datasets reveals human cis-regulatory regions Hamid Bolouri & Larry Ruzzo Thanks to Stephen Tapscott, Steven Henikoff & Zizhen Yao Slides will be available from: http://labs.fhcrc.org/bolouri/ Email [email protected] for manuscript (Bolouri & Ruzzo, submitted) Kleinjan & van Heyningen, Am. J. Hum. Genet., 2005, (76)8–32 Epstein, Briefings in Func. Genom. & Protemoics, 2009, 8(4)310-16 Regulation of SPi1 (Sfpi1, PU.1 protein) expression – part 1 miR155*, miR569# ~750nt promoter ~250nt promoter The antisense RNA • causes translational stalling • has its own promoter • requires distal SPI1 enhancer • is transcribed with/without SPI1. # Hikami et al, Arthritis & Rheumatism, 2011, 63(3):755–763 * Vigorito et al, 2007, Immunity 27, 847–859 Ebralidze et al, Genes & Development, 2008, 22: 2085-2092. Regulation of SPi1 expression – part 2 (mouse coordinates) Bidirectional ncRNA transcription proportional to PU.1 expression PU.1/ELF1/FLI1/GLI1 GATA1 GATA1 Sox4/TCF/LEF PU.1 RUNX1 SP1 RUNX1 RUNX1 SP1 ELF1 NF-kB SATB1 IKAROS PU.1 cJun/CEBP OCT1 cJun/CEBP 500b 500b 500b 500b 500b 750b 500b -18Kb -14Kb -12Kb -10Kb -9Kb Chou et al, Blood, 2009, 114: 983-994 Hoogenkamp et al, Molecular & Cellular Biology, 2007, 27(21):7425-7438 Zarnegar & Rothenberg, 2010, Mol. & cell Biol. 4922-4939 An NF-kB binding-site variant in the SPI1 URE reduces PU.1 expression & is GGGCCTCCCC correlated with AML GGGTCTTCCC Bonadies et al, Oncogene, 2009, 29(7):1062-72. SATB1 binding site A distal single nucleotide polymorphism alters long- range regulation of the PU.1 gene in acute myeloid leukemia Steidl et al, J Clin Invest. -

Integrating Protein Copy Numbers with Interaction Networks to Quantify Stoichiometry in Mammalian Endocytosis
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Integrating protein copy numbers with interaction networks to quantify stoichiometry in mammalian endocytosis Daisy Duan1, Meretta Hanson1, David O. Holland2, Margaret E Johnson1* 1TC Jenkins Department of Biophysics, Johns Hopkins University, 3400 N Charles St, Baltimore, MD 21218. 2NIH, Bethesda, MD, 20892. *Corresponding Author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.10.29.361196; this version posted October 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Proteins that drive processes like clathrin-mediated endocytosis (CME) are expressed at various copy numbers within a cell, from hundreds (e.g. auxilin) to millions (e.g. clathrin). Between cell types with identical genomes, copy numbers further vary significantly both in absolute and relative abundance. These variations contain essential information about each protein’s function, but how significant are these variations and how can they be quantified to infer useful functional behavior? Here, we address this by quantifying the stoichiometry of proteins involved in the CME network. We find robust trends across three cell types in proteins that are sub- vs super-stoichiometric in terms of protein function, network topology (e.g. -

Figure S1. HAEC ROS Production and ML090 NOX5-Inhibition
Figure S1. HAEC ROS production and ML090 NOX5-inhibition. (a) Extracellular H2O2 production in HAEC treated with ML090 at different concentrations and 24 h after being infected with GFP and NOX5-β adenoviruses (MOI 100). **p< 0.01, and ****p< 0.0001 vs control NOX5-β-infected cells (ML090, 0 nM). Results expressed as mean ± SEM. Fold increase vs GFP-infected cells with 0 nM of ML090. n= 6. (b) NOX5-β overexpression and DHE oxidation in HAEC. Representative images from three experiments are shown. Intracellular superoxide anion production of HAEC 24 h after infection with GFP and NOX5-β adenoviruses at different MOIs treated or not with ML090 (10 nM). MOI: Multiplicity of infection. Figure S2. Ontology analysis of HAEC infected with NOX5-β. Ontology analysis shows that the response to unfolded protein is the most relevant. Figure S3. UPR mRNA expression in heart of infarcted transgenic mice. n= 12-13. Results expressed as mean ± SEM. Table S1: Altered gene expression due to NOX5-β expression at 12 h (bold, highlighted in yellow). N12hvsG12h N18hvsG18h N24hvsG24h GeneName GeneDescription TranscriptID logFC p-value logFC p-value logFC p-value family with sequence similarity NM_052966 1.45 1.20E-17 2.44 3.27E-19 2.96 6.24E-21 FAM129A 129. member A DnaJ (Hsp40) homolog. NM_001130182 2.19 9.83E-20 2.94 2.90E-19 3.01 1.68E-19 DNAJA4 subfamily A. member 4 phorbol-12-myristate-13-acetate- NM_021127 0.93 1.84E-12 2.41 1.32E-17 2.69 1.43E-18 PMAIP1 induced protein 1 E2F7 E2F transcription factor 7 NM_203394 0.71 8.35E-11 2.20 2.21E-17 2.48 1.84E-18 DnaJ (Hsp40) homolog. -

Virtual Chip-Seq: Predicting Transcription Factor Binding
bioRxiv preprint doi: https://doi.org/10.1101/168419; this version posted March 12, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Virtual ChIP-seq: predicting transcription factor binding 2 by learning from the transcriptome 1,2,3 1,2,3,4,5 3 Mehran Karimzadeh and Michael M. Hoffman 1 4 Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada 2 5 Princess Margaret Cancer Centre, Toronto, ON, Canada 3 6 Vector Institute, Toronto, ON, Canada 4 7 Department of Computer Science, University of Toronto, Toronto, ON, Canada 5 8 Lead contact: michael.hoff[email protected] 9 March 8, 2019 10 Abstract 11 Motivation: 12 Identifying transcription factor binding sites is the first step in pinpointing non-coding mutations 13 that disrupt the regulatory function of transcription factors and promote disease. ChIP-seq is 14 the most common method for identifying binding sites, but performing it on patient samples is 15 hampered by the amount of available biological material and the cost of the experiment. Existing 16 methods for computational prediction of regulatory elements primarily predict binding in genomic 17 regions with sequence similarity to known transcription factor sequence preferences. This has limited 18 efficacy since most binding sites do not resemble known transcription factor sequence motifs, and 19 many transcription factors are not even sequence-specific. 20 Results: 21 We developed Virtual ChIP-seq, which predicts binding of individual transcription factors in new 22 cell types using an artificial neural network that integrates ChIP-seq results from other cell types 23 and chromatin accessibility data in the new cell type.