Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Core Transcriptional Regulatory Circuitries in Cancer
Oncogene (2020) 39:6633–6646 https://doi.org/10.1038/s41388-020-01459-w REVIEW ARTICLE Core transcriptional regulatory circuitries in cancer 1 1,2,3 1 2 1,4,5 Ye Chen ● Liang Xu ● Ruby Yu-Tong Lin ● Markus Müschen ● H. Phillip Koeffler Received: 14 June 2020 / Revised: 30 August 2020 / Accepted: 4 September 2020 / Published online: 17 September 2020 © The Author(s) 2020. This article is published with open access Abstract Transcription factors (TFs) coordinate the on-and-off states of gene expression typically in a combinatorial fashion. Studies from embryonic stem cells and other cell types have revealed that a clique of self-regulated core TFs control cell identity and cell state. These core TFs form interconnected feed-forward transcriptional loops to establish and reinforce the cell-type- specific gene-expression program; the ensemble of core TFs and their regulatory loops constitutes core transcriptional regulatory circuitry (CRC). Here, we summarize recent progress in computational reconstitution and biologic exploration of CRCs across various human malignancies, and consolidate the strategy and methodology for CRC discovery. We also discuss the genetic basis and therapeutic vulnerability of CRC, and highlight new frontiers and future efforts for the study of CRC in cancer. Knowledge of CRC in cancer is fundamental to understanding cancer-specific transcriptional addiction, and should provide important insight to both pathobiology and therapeutics. 1234567890();,: 1234567890();,: Introduction genes. Till now, one critical goal in biology remains to understand the composition and hierarchy of transcriptional Transcriptional regulation is one of the fundamental mole- regulatory network in each specified cell type/lineage. -

Cellular Responses to Erbb-2 Overexpression in Human Mammary Luminal Epithelial Cells: Comparison of Mrna and Protein Expression
British Journal of Cancer (2004) 90, 173 – 181 & 2004 Cancer Research UK All rights reserved 0007 – 0920/04 $25.00 www.bjcancer.com Cellular responses to ErbB-2 overexpression in human mammary luminal epithelial cells: comparison of mRNA and protein expression SL White1, S Gharbi1, MF Bertani1, H-L Chan1, MD Waterfield1 and JF Timms*,1 1 Ludwig Institute for Cancer Research, Wing 1.1, Cruciform Building, Gower Street, London WCIE 6BT, UK Microarray analysis offers a powerful tool for studying the mechanisms of cellular transformation, although the correlation between mRNA and protein expression is largely unknown. In this study, a microarray analysis was performed to compare transcription in response to overexpression of the ErbB-2 receptor tyrosine kinase in a model mammary luminal epithelial cell system, and in response to the ErbB-specific growth factor heregulin b1. We sought to validate mRNA changes by monitoring changes at the protein level using a parallel proteomics strategy, and report a surprisingly high correlation between transcription and translation for the subset of genes studied. We further characterised the identified targets and relate differential expression to changes in the biological properties of ErbB-2-overexpressing cells. We found differential regulation of several key cell cycle modulators, including cyclin D2, and downregulation of a large number of interferon-inducible genes, consistent with increased proliferation of the ErbB-2- overexpressing cells. Furthermore, differential expression of genes involved in extracellular matrix modelling and cellular adhesion was linked to altered adhesion of these cells. Finally, we provide evidence for enhanced autocrine activation of MAPK signalling and the AP-1 transcription complex. -

An Order Estimation Based Approach to Identify Response Genes
AN ORDER ESTIMATION BASED APPROACH TO IDENTIFY RESPONSE GENES FOR MICRO ARRAY TIME COURSE DATA A Thesis Presented to The Faculty of Graduate Studies of The University of Guelph by ZHIHENG LU In partial fulfilment of requirements for the degree of Doctor of Philosophy September, 2008 © Zhiheng Lu, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-47605-5 Our file Notre reference ISBN: 978-0-494-47605-5 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -
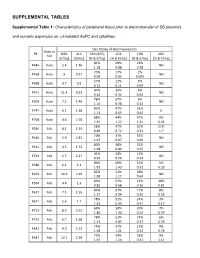
Supplemental Tables
SUPPLEMENTAL TABLES Supplemental Table 1: Characteristics of peripheral blood prior to electrotransfer of SB plasmids and numeric expansion on γ-irradiated AaPC and cytokines Day 0 (Day of electroporation) Auto or P# WBC ALC CD3 (ATC) CD4 CD8 ABC Allo [K/mL] [K/mL] [% & K/mL] [% & K/mL] [% & K/mL] [% & K/mL] 81% 60% 19% P446 Auto 5.4 1.46 ND 1.18 0.88 0.28 23% 17% 2% P458 Auto 9 0.27 ND 0.06 0.05 0.005 17% 12% 5% P468 Auto 4.7 0.9 ND 0.15 0.11 0.04 47% 35% 5% P471 Auto 11.4 0.93 ND 0.44 0.32 0.04 78% 67% 9% P509 Auto 7.2 1.46 ND 1.14 0.98 0.13 82% 47% 32% P747 Auto 6.1 1.38 0 1.13 0.65 0.44 88% 44% 47% 0% P708 Auto 4.6 2.78 2.45 1.22 1.31 0.05 58% 47% 20% 21% P396 Allo 8.1 1.54 0.89 0.72 0.31 1.7 70% 31% 32% P410 Allo 5.9 2.81 ND 1.97 0.87 0.90 80% 48% 32% P411 Allo 4.5 1.72 ND 1.38 0.83 0.55 41% 24% 15% P513 Allo 6.7 2.27 ND 0.93 0.54 0.34 86% 68% 15% 5% P580 Allo 6.1 2.1 1.81 1.43 0.32 0.29 82% 51% 28% P459 Allo 10.6 2.29 ND 1.88 1.17 0.64 64% 52% 12% 18% P564 Allo 4.4 1.3 0.83 0.68 0.16 0.81 82% 61% 17% 8% P617 Allo 7.1 1.55 1.27 0.94 0.26 0.58 78% 53% 24% 3% P671 Allo 5.4 1.7 1.33 0.90 0.41 0.17 69% 50% 20% 7% P723 Allo 8.6 2.61 1.80 1.30 0.52 0.59 78% 52% 22% 6% P732 Allo 6.7 1.68 1.31 0.87 0.37 0.39 74% 57% 15% 9% P641 Allo 9.0 2.13 1.58 1.21 0.32 0.78 73% 54% 18% 9% P647 Allo 12.1 2.29 1.67 1.24 0.41 1.11 75% 59% 11% P716 Allo 3.0 1.83 0 1.37 1.08 0.20 87% 73% 12% 1% P718 Allo 6.2 1.99 1.73 1.45 0.24 0.07 83% 69% 13% 6% P771 Allo 13.1 3.23 2.68 2.23 0.42 0.79 73% 42% 27% 6% P783 Allo 8.1 1.54 1.12 0.65 0.42 0.52 WBC = white blood -

Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug
cancers Article Harnessing Gene Expression Profiles for the Identification of Ex Vivo Drug Response Genes in Pediatric Acute Myeloid Leukemia David G.J. Cucchi 1 , Costa Bachas 1 , Marry M. van den Heuvel-Eibrink 2,3, Susan T.C.J.M. Arentsen-Peters 3, Zinia J. Kwidama 1, Gerrit J. Schuurhuis 1, Yehuda G. Assaraf 4, Valérie de Haas 3 , Gertjan J.L. Kaspers 3,5 and Jacqueline Cloos 1,* 1 Hematology, Cancer Center Amsterdam, Amsterdam UMC, Vrije Universiteit Amsterdam, 1081 HV Amsterdam, The Netherlands; [email protected] (D.G.J.C.); [email protected] (C.B.); [email protected] (Z.J.K.); [email protected] (G.J.S.) 2 Department of Pediatric Oncology/Hematology, Erasmus MC–Sophia Children’s Hospital, 3015 CN Rotterdam, The Netherlands; [email protected] 3 Princess Máxima Center for Pediatric Oncology, 3584 CS Utrecht, The Netherlands; [email protected] (S.T.C.J.M.A.-P.); [email protected] (V.d.H.); [email protected] (G.J.L.K.) 4 The Fred Wyszkowski Cancer Research, Laboratory, Department of Biology, Technion-Israel Institute of Technology, 3200003 Haifa, Israel; [email protected] 5 Emma’s Children’s Hospital, Amsterdam UMC, Vrije Universiteit Amsterdam, Pediatric Oncology, 1081 HV Amsterdam, The Netherlands * Correspondence: [email protected] Received: 21 April 2020; Accepted: 12 May 2020; Published: 15 May 2020 Abstract: Novel treatment strategies are of paramount importance to improve clinical outcomes in pediatric AML. Since chemotherapy is likely to remain the cornerstone of curative treatment of AML, insights in the molecular mechanisms that determine its cytotoxic effects could aid further treatment optimization. -

Genome-Wide Inference of Natural Selection on Human Transcription Factor Binding Sites
ANALYSIS Genome-wide inference of natural selection on human transcription factor binding sites Leonardo Arbiza1, Ilan Gronau1, Bulent A Aksoy2, Melissa J Hubisz1, Brad Gulko3, Alon Keinan1–3 & Adam Siepel1–3 For decades, it has been hypothesized that gene regulation persistence in humans7,8. In addition, some genome-wide analyses has had a central role in human evolution, yet much remains have found bulk statistical evidence of natural selection in noncoding unknown about the genome-wide impact of regulatory regions near genes, presumably due to cis-regulatory elements9–12. mutations. Here we use whole-genome sequences and genome- Nevertheless, evidence in support of the overall prominence of wide chromatin immunoprecipitation and sequencing data to cis-regulatory mutations in evolutionary adaptation remains largely demonstrate that natural selection has profoundly influenced anecdotal and indirect, and there is continuing controversy about the human transcription factor binding sites since the divergence relative roles of regulatory and protein-coding sequences in evolu- of humans from chimpanzees 4–6 million years ago. Our tion8. Large-scale genomic studies of the evolution of transcription analysis uses a new probabilistic method, called INSIGHT, for factor binding sites have the potential to advance this debate, but a measuring the influence of selection on collections of short, major limitation of such studies so far has been a lack of precisely interspersed noncoding elements. We find that, on average, annotated binding sites across the genome. The analysis of con- transcription factor binding sites have experienced somewhat served noncoding sequences and/or promoter regions rather than weaker selection than protein-coding genes. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Gene Expression Profile in Different Age Groups and Its Association With
cells Article Gene Expression Profile in Different Age Groups and Its Association with Cognitive Function in Healthy Malay Adults in Malaysia Nur Fathiah Abdul Sani 1 , Ahmad Imran Zaydi Amir Hamzah 1, Zulzikry Hafiz Abu Bakar 1 , Yasmin Anum Mohd Yusof 2, Suzana Makpol 1 , Wan Zurinah Wan Ngah 1 and Hanafi Ahmad Damanhuri 1,* 1 Department of Biochemistry, Faculty of Medicine, Universiti Kebangsaan Malaysia Medical Center, Jalan Yaacob Latif, Cheras, Kuala Lumpur 56000, Malaysia; [email protected] (N.F.A.S.); [email protected] (A.I.Z.A.H.); zulzikryhafi[email protected] (Z.H.A.B.); [email protected] (S.M.); [email protected] (W.Z.W.N.) 2 Faculty of Medicine and Defence Health, National Defence University of Malaysia, Kem Sungai Besi, Kuala Lumpur 57000, Malaysia; [email protected] * Correspondence: hanafi[email protected] Abstract: The mechanism of cognitive aging at the molecular level is complex and not well under- stood. Growing evidence suggests that cognitive differences might also be caused by ethnicity. Thus, this study aims to determine the gene expression changes associated with age-related cognitive decline among Malay adults in Malaysia. A cross-sectional study was conducted on 160 healthy Malay subjects, aged between 28 and 79, and recruited around Selangor and Klang Valley, Malaysia. Citation: Abdul Sani, N.F.; Amir Gene expression analysis was performed using a HumanHT-12v4.0 Expression BeadChip microarray Hamzah, A.I.Z.; Abu Bakar, Z.H.; kit. The top 20 differentially expressed genes at p < 0.05 and fold change (FC) = 1.2 showed that Mohd Yusof, Y.A.; Makpol, S.; Wan PAFAH1B3, HIST1H1E, KCNA3, TM7SF2, RGS1, and TGFBRAP1 were regulated with increased Ngah, W.Z.; Damanhuri, H.A. -

Functional Characterization of the New 8Q21 Asthma Risk Locus
Functional characterization of the new 8q21 Asthma risk locus Cristina M T Vicente B.Sc, M.Sc A thesis submitted for the degree of Doctor of Philosophy at The University of Queensland in 2017 Faculty of Medicine Abstract Genome wide association studies (GWAS) provide a powerful tool to identify genetic variants associated with asthma risk. However, the target genes for many allergy risk variants discovered to date are unknown. In a recent GWAS, Ferreira et al. identified a new association between asthma risk and common variants located on chromosome 8q21. The overarching aim of this thesis was to elucidate the biological mechanisms underlying this association. Specifically, the goals of this study were to identify the gene(s) underlying the observed association and to study their contribution to asthma pathophysiology. Using genetic data from the 1000 Genomes Project, we first identified 118 variants in linkage disequilibrium (LD; r2>0.6) with the sentinel allergy risk SNP (rs7009110) on chromosome 8q21. Of these, 35 were found to overlap one of four Putative Regulatory Elements (PREs) identified in this region in a lymphoblastoid cell line (LCL), based on epigenetic marks measured by the ENCODE project. Results from analysis of gene expression data generated for LCLs (n=373) by the Geuvadis consortium indicated that rs7009110 is associated with the expression of only one nearby gene: PAG1 - located 732 kb away. PAG1 encodes a transmembrane adaptor protein localized to lipid rafts, which is highly expressed in immune cells. Results from chromosome conformation capture (3C) experiments showed that PREs in the region of association physically interacted with the promoter of PAG1. -
![Downloaded from JASPAR (JASPAR ID: MA0527.1) Across All Identified Peaks Using the Matrix-Scan Module from RSAT [20]](https://docslib.b-cdn.net/cover/5028/downloaded-from-jaspar-jaspar-id-ma0527-1-across-all-identified-peaks-using-the-matrix-scan-module-from-rsat-20-355028.webp)
Downloaded from JASPAR (JASPAR ID: MA0527.1) Across All Identified Peaks Using the Matrix-Scan Module from RSAT [20]
bioRxiv preprint doi: https://doi.org/10.1101/585653; this version posted March 24, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ZBTB33 (Kaiso) methylated binding sites are associated with primed heterochromatin Quy Xiao Xuan Lin1, Khadija Rebbani1, Sudhakar Jha1,2, Touati Benoukraf1,3* 1Cancer Science Institute of Singapore, National University of Singapore, Singapore, Singapore 2Department of Biochemistry, National University of Singapore, Singapore, Singapore 3Discipline of Genetics, Faculty of Medicine, Memorial University of Newfoundland, St. John’s, NL, Canada *Correspondence to: Touati Benoukraf, Ph.D. Faculty of Medicine, Discipline of Genetics Cancer Science Institute of Singapore Craig L. Dobbin Genetics Research Centre National University of Singapore Room 5M317 Centre for Translational Medicine, Memorial University of Newfoundland 14 Medical Drive, #12-01 St. John's, NL A1B 3V6 Singapore 117599 Canada Phone: +1 (709) 864-6671 Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/585653; this version posted March 24, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract Background: ZBTB33, also known as Kaiso, is a member of the zinc finger and BTB/POZ family. In contrast to many transcription factors, ZBTB33 has the ability to bind both a sequence-specific consensus and methylated DNA. -

UNIVERSITY of CALIFORNIA, IRVINE Combinatorial Regulation By
UNIVERSITY OF CALIFORNIA, IRVINE Combinatorial regulation by maternal transcription factors during activation of the endoderm gene regulatory network DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biological Sciences by Kitt D. Paraiso Dissertation Committee: Professor Ken W.Y. Cho, Chair Associate Professor Olivier Cinquin Professor Thomas Schilling 2018 Chapter 4 © 2017 Elsevier Ltd. © 2018 Kitt D. Paraiso DEDICATION To the incredibly intelligent and talented people, who in one way or another, helped complete this thesis. ii TABLE OF CONTENTS Page LIST OF FIGURES vii LIST OF TABLES ix LIST OF ABBREVIATIONS X ACKNOWLEDGEMENTS xi CURRICULUM VITAE xii ABSTRACT OF THE DISSERTATION xiv CHAPTER 1: Maternal transcription factors during early endoderm formation in 1 Xenopus Transcription factors co-regulate in a cell type-specific manner 2 Otx1 is expressed in a variety of cell lineages 4 Maternal otx1 in the endodermal conteXt 5 Establishment of enhancers by maternal transcription factors 9 Uncovering the endodermal gene regulatory network 12 Zygotic genome activation and temporal control of gene eXpression 14 The role of maternal transcription factors in early development 18 References 19 CHAPTER 2: Assembly of maternal transcription factors initiates the emergence 26 of tissue-specific zygotic cis-regulatory regions Introduction 28 Identification of maternal vegetally-localized transcription factors 31 Vegt and OtX1 combinatorially regulate the endodermal 33 transcriptome iii -

P53 — 30 Yreviewsears On
FOCUS ON P53 — 30 YREVIEWSEARS ON p53 — a Jack of all trades but master of none Melissa R. Junttila and Gerard I. Evan Abstract | Cancers are rare because their evolution is actively restrained by a range of tumour suppressors. Of these p53 seems unusually crucial as either it or its attendant upstream or downstream pathways are inactivated in virtually all cancers. p53 is an evolutionarily ancient coordinator of metazoan stress responses. Its role in tumour suppression is likely to be a relatively recent adaptation, which is only necessary when large, long-lived organisms acquired the sufficient size and somatic regenerative capacity to necessitate specific mechanisms to reign in rogue proliferating cells. However, such evolutionary reappropriation of this venerable transcription factor entails compromises that restrict its efficacy as a tumour suppressor. Cnidarians–bilaterians Cancer is a genetic pathology that arises in the adult tissues questions are important for several reasons. Knowing Cinidarians comprise an animal of long-lived organisms, such as vertebrates, whose tis- how p53 discriminates between normal and tumour cells phylum of ~9,000 radially sues retain a substantial regenerative capacity throughout might point to attributes of cancer cells that qualitatively symmetrical, mostly marine life and, consequently, whose somatic cells accumulate distinguish them from normal somatic cells and could organisms. Most other animals are bilaterally symmetrical mutations. Occasionally, such mutations corrupt the be used as tumour-specific targets. Knowing what sig- and are classed as bilateria. regulatory mechanisms that suppress untoward somatic nals drive selection for loss of p53 function in cancers The cnidarians and bilaterians cell proliferation, survival and migration, resulting in the would help us understand what constrains and dictates last shared a common ancestor progressive outgrowth of a somatic clone.