P53 — 30 Yreviewsears On
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Order Estimation Based Approach to Identify Response Genes
AN ORDER ESTIMATION BASED APPROACH TO IDENTIFY RESPONSE GENES FOR MICRO ARRAY TIME COURSE DATA A Thesis Presented to The Faculty of Graduate Studies of The University of Guelph by ZHIHENG LU In partial fulfilment of requirements for the degree of Doctor of Philosophy September, 2008 © Zhiheng Lu, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-47605-5 Our file Notre reference ISBN: 978-0-494-47605-5 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -
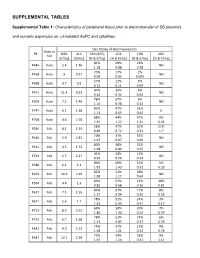
Supplemental Tables
SUPPLEMENTAL TABLES Supplemental Table 1: Characteristics of peripheral blood prior to electrotransfer of SB plasmids and numeric expansion on γ-irradiated AaPC and cytokines Day 0 (Day of electroporation) Auto or P# WBC ALC CD3 (ATC) CD4 CD8 ABC Allo [K/mL] [K/mL] [% & K/mL] [% & K/mL] [% & K/mL] [% & K/mL] 81% 60% 19% P446 Auto 5.4 1.46 ND 1.18 0.88 0.28 23% 17% 2% P458 Auto 9 0.27 ND 0.06 0.05 0.005 17% 12% 5% P468 Auto 4.7 0.9 ND 0.15 0.11 0.04 47% 35% 5% P471 Auto 11.4 0.93 ND 0.44 0.32 0.04 78% 67% 9% P509 Auto 7.2 1.46 ND 1.14 0.98 0.13 82% 47% 32% P747 Auto 6.1 1.38 0 1.13 0.65 0.44 88% 44% 47% 0% P708 Auto 4.6 2.78 2.45 1.22 1.31 0.05 58% 47% 20% 21% P396 Allo 8.1 1.54 0.89 0.72 0.31 1.7 70% 31% 32% P410 Allo 5.9 2.81 ND 1.97 0.87 0.90 80% 48% 32% P411 Allo 4.5 1.72 ND 1.38 0.83 0.55 41% 24% 15% P513 Allo 6.7 2.27 ND 0.93 0.54 0.34 86% 68% 15% 5% P580 Allo 6.1 2.1 1.81 1.43 0.32 0.29 82% 51% 28% P459 Allo 10.6 2.29 ND 1.88 1.17 0.64 64% 52% 12% 18% P564 Allo 4.4 1.3 0.83 0.68 0.16 0.81 82% 61% 17% 8% P617 Allo 7.1 1.55 1.27 0.94 0.26 0.58 78% 53% 24% 3% P671 Allo 5.4 1.7 1.33 0.90 0.41 0.17 69% 50% 20% 7% P723 Allo 8.6 2.61 1.80 1.30 0.52 0.59 78% 52% 22% 6% P732 Allo 6.7 1.68 1.31 0.87 0.37 0.39 74% 57% 15% 9% P641 Allo 9.0 2.13 1.58 1.21 0.32 0.78 73% 54% 18% 9% P647 Allo 12.1 2.29 1.67 1.24 0.41 1.11 75% 59% 11% P716 Allo 3.0 1.83 0 1.37 1.08 0.20 87% 73% 12% 1% P718 Allo 6.2 1.99 1.73 1.45 0.24 0.07 83% 69% 13% 6% P771 Allo 13.1 3.23 2.68 2.23 0.42 0.79 73% 42% 27% 6% P783 Allo 8.1 1.54 1.12 0.65 0.42 0.52 WBC = white blood -

Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Integration of 198 ChIP-seq datasets reveals human cis-regulatory regions Hamid Bolouri & Larry Ruzzo Thanks to Stephen Tapscott, Steven Henikoff & Zizhen Yao Slides will be available from: http://labs.fhcrc.org/bolouri/ Email [email protected] for manuscript (Bolouri & Ruzzo, submitted) Kleinjan & van Heyningen, Am. J. Hum. Genet., 2005, (76)8–32 Epstein, Briefings in Func. Genom. & Protemoics, 2009, 8(4)310-16 Regulation of SPi1 (Sfpi1, PU.1 protein) expression – part 1 miR155*, miR569# ~750nt promoter ~250nt promoter The antisense RNA • causes translational stalling • has its own promoter • requires distal SPI1 enhancer • is transcribed with/without SPI1. # Hikami et al, Arthritis & Rheumatism, 2011, 63(3):755–763 * Vigorito et al, 2007, Immunity 27, 847–859 Ebralidze et al, Genes & Development, 2008, 22: 2085-2092. Regulation of SPi1 expression – part 2 (mouse coordinates) Bidirectional ncRNA transcription proportional to PU.1 expression PU.1/ELF1/FLI1/GLI1 GATA1 GATA1 Sox4/TCF/LEF PU.1 RUNX1 SP1 RUNX1 RUNX1 SP1 ELF1 NF-kB SATB1 IKAROS PU.1 cJun/CEBP OCT1 cJun/CEBP 500b 500b 500b 500b 500b 750b 500b -18Kb -14Kb -12Kb -10Kb -9Kb Chou et al, Blood, 2009, 114: 983-994 Hoogenkamp et al, Molecular & Cellular Biology, 2007, 27(21):7425-7438 Zarnegar & Rothenberg, 2010, Mol. & cell Biol. 4922-4939 An NF-kB binding-site variant in the SPI1 URE reduces PU.1 expression & is GGGCCTCCCC correlated with AML GGGTCTTCCC Bonadies et al, Oncogene, 2009, 29(7):1062-72. SATB1 binding site A distal single nucleotide polymorphism alters long- range regulation of the PU.1 gene in acute myeloid leukemia Steidl et al, J Clin Invest. -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Methodology article Open Access "NeuroStem Chip": a novel highly specialized tool to study neural differentiation pathways in human stem cells Sergey V Anisimov*, Nicolaj S Christophersen, Ana S Correia, Jia-Yi Li and Patrik Brundin Address: Neuronal Survival Unit, Wallenberg Neuroscience Center, Lund University, 221 84 Lund, Sweden Email: Sergey V Anisimov* - [email protected]; Nicolaj S Christophersen - [email protected]; Ana S Correia - [email protected]; Jia-Yi Li - [email protected]; Patrik Brundin - [email protected] * Corresponding author Published: 8 February 2007 Received: 21 September 2006 Accepted: 8 February 2007 BMC Genomics 2007, 8:46 doi:10.1186/1471-2164-8-46 This article is available from: http://www.biomedcentral.com/1471-2164/8/46 © 2007 Anisimov et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Human stem cells are viewed as a possible source of neurons for a cell-based therapy of neurodegenerative disorders, such as Parkinson's disease. Several protocols that generate different types of neurons from human stem cells (hSCs) have been developed. Nevertheless, the cellular mechanisms that underlie the development of neurons in vitro as they are subjected to the specific differentiation protocols are often poorly understood. Results: We have designed a focused DNA (oligonucleotide-based) large-scale microarray platform (named "NeuroStem Chip") and used it to study gene expression patterns in hSCs as they differentiate into neurons. -

Prostate News
Prostate Cancer and Prostatic Diseases (2005) 8, 2–6 & 2005 Nature Publishing Group All rights reserved 1365-7852/05 $30.00 www.nature.com/pcan Research Highlights Prostate News Prostate Cancer and Prostatic Diseases (2005) 8, 2–6. doi:10.1038/sj.pcan.4500793 Pokemon’s central role in cancer progression of prostate cancer. Epigenetic changes are reversible, which make them attractive targets for cancer development treatment with modulators that demethylate DNA and Researchers at New York’s Memorial Sloan-Kettering inhibit histone deacetylases, leading to reactivation of Cancer Center report novel findings in Nature regarding silenced genes. DNA methylation and histone modifica- a new cellular oncogene implicated in the development tions are important epigenetic mechanisms of gene of human cancers, including prostate neoplasia. Poke- regulation, and both play essential roles in tumor mon, a pop-culture inspired acronym for ‘POK erythroid initiation and progression. In prostate cancer, aberrant myeloid ontogenic factor’, is a POK protein family epigenetic events such as DNA hypo- and hypermethy- member encoded by the ZbTb7 gene and characterized lation and altered histone acetylation have been by its critical transcription function in cellular differ- observed to affect a large number of genes. However, entiation. Takahiro Maeda, Pier Paolo Pandolfi and no plenitude is without peril: although the number of colleagues show that Pokemon acts as a genuine proto- genes that undergo aberrant epigenetic inactivation oncogene when overexpressed by inhibiting the expres- associated with prostate cancer seems to be growing, sion of tumor suppressors, such as ARF. very few of these genes have yielded promising results To establish this novel finding, the authors conducted as potential tumor biomarkers for early diagnosis and a complete battery of genetic, clinical, and pathological risk assessment. -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: • This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. • A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. • This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. • The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. • When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Regulation and Function of miR-199-3p in Murine and Human Cytomegalovirus Infections Nouf N. M. Laqtom s0898296 A thesis submitted in fulfilment of requirements for the degree of Doctor of Philosophy Division of Pathway Medicine, School of Biomedical Sciences The University of Edinburgh May 2013 Supervisors: Dr. Amy Buck and Dr. Bernadette Dutia i Declaration of Authorship I hereby declare that this thesis is of my own composition, and that it contains no material previously submitted for the award of any other degree. The work reported in this thesis has been executed by myself, except where due acknowledgement is made in the text. Nouf N. M. Laqtom ii Abstract Human Cytomegalovirus (HCMV), the prototypic β-herpesvirus, is the most common cause of congenital infections as well as morbidity and mortality in immunocompromised patients. -

Establishment of Mouse Primed Stem Cells by Combination of Activin and LIF Signaling
fcell-09-713503 July 31, 2021 Time: 12:43 # 1 ORIGINAL RESEARCH published: 05 August 2021 doi: 10.3389/fcell.2021.713503 Establishment of Mouse Primed Stem Cells by Combination of Activin and LIF Signaling Mengyi Wei1,2†, Yanglin Chen1,2,3†, Chaoyue Zhao1,2, Li Zheng1,2, Baojiang Wu1,2, Chen Chen1,2, Xihe Li1,2,4 and Siqin Bao1,2* 1 State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, Inner Mongolia University, Hohhot, China, 2 Institute of Animal Genetic Research of Mongolia Plateau, College of Life Sciences, Inner Mongolia University, Hohhot, China, 3 School of Basic Medical Sciences, Southern Medical University, Guangzhou, China, 4 Inner Mongolia Saikexing Institute of Breeding and Reproductive Biotechnology in Domestic Animal, Hohhot, China In mice, embryonic stem cells (ESCs) and epiblast stem cells (EpiSCs) are established from pre- and post-implantation embryos and represent the naive and primed state, respectively. Herein we used mouse leukemia inhibitory factor (LIF), which supports Edited by: Wei Jiang, ESCs self-renewal and Activin A (Act A), which is the main factor in maintaining EpiSCs Wuhan University, China in post-implantation epiblast cultures, to derive a primed stem cell line named ALSCs. Reviewed by: Like EpiSCs, ALSCs express key pluripotent genes Oct4, Sox2, and Nanog; one X Tetsuya S. Tanaka, Elixirgen Scientific, Inc., United States chromosome was inactivated; and the cells failed to contribute to chimera formation Fabiana Passaro, in vivo. Notably, compared to EpiSCs, ALSCs efficiently reversed to ESCs (rESCs) University of Naples Federico II, Italy on activation of Wnt signaling. Moreover, we also discovered that culturing EpiSCs in *Correspondence: AL medium for several passages favored Wnt signaling-driven naive pluripotency. -

Zbtb7a and Zbtb7b: Opening Naïve Loci to Reprogram Escs
BioScience Trends. 2021; 15(1):58-60. 58 BioScience Trends. 2021; 15(1):58-60. Letter DOI: 10.5582/bst.2020.03429 Zbtb7a and Zbtb7b: Opening naïve loci to reprogram ESCs Hua Cao1, Hong Huang2,3,*, Huifang Tang1,2,* 1 Department of Cardiology, The First Affiliated Hospital of University of South China, Hengyang, Hunan, China; 2 Institute of Cardiovascular Disease, The First Affiliated Hospital of University of South China, Hengyang, Hunan, China; 3 Institute of Clinical Medicine, The First Affiliated Hospital of University of South China, Hengyang, Hunan Province, China. SUMMARY Bone morphogenetic protein 4 (BMP4) was recently reported to confer reprogramming capability to embryonic stem cells (ESCs) by reactivating naïve pluripotency genes via Zbtb7a and Zbtb7b. A visual reporting system was developed to first identify BMP4 as a driver for the primed-to-naïve transition (PNT). In addition, two specific inhibitors were identified as significantly improving the efficiency of PNT (~80% transition) within 8 days. The Zbtb7 family members were first introduced in the context of PNT and stem cell fate decision-making. These findings provide valuable information on acquiring naïve pluripotent stem cells for regenerative medicine. Keywords BMP4, fate decision-making, PNT, reprogramming The primed-to-naïve transition (PNT) between embryonic contribute to increase DNA methylation and to reduce stem cells (ESCs) and epiblast stem cells (EpiSCs) is chromatin accessibility (3). Gene expression is closely finely tuned by a set of transcription factors (TFs) and linked to chromatin accessibility, so whether BMP4 chemicals (1). Naïve pluripotency is defined as the self- regulates chromatin interaction during PNT has yet to be renewal stage of ESCs, which gives rise to a whole determined. -

Product Datasheet ZBTB7A/Pokemon Antibody
Product Datasheet ZBTB7A/Pokemon Antibody NB100-761 Unit Size: 100 ul Store at 4C. Do not freeze. Publications: 4 Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NB100-761 Updated 2/9/2021 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NB100-761 Page 1 of 3 v.20.1 Updated 2/9/2021 NB100-761 ZBTB7A/Pokemon Antibody Product Information Unit Size 100 ul Concentration 1.0 mg/ml Storage Store at 4C. Do not freeze. Clonality Polyclonal Preservative 0.09% Sodium Azide Isotype IgG Purity Immunogen affinity purified Buffer Tris-Citrate/Phosphate (pH 7.0 - 8.0) Product Description Host Rabbit Gene ID 51341 Gene Symbol ZBTB7A Species Human, Mouse Immunogen The immunogen recognized by this antibody maps to a region between residues 200 and 250 of human ZBTB7 (Factor Binding IST protein 1) using the numbering given in entry NP_056982.1 (GeneID 51341). Product Application Details Applications Western Blot, Immunohistochemistry, Immunoprecipitation Recommended Dilutions Western Blot 1:3000-1:15000, Immunohistochemistry, Immunoprecipitation 2-5 ug/mg lysate Application Notes IHC reactivity reported in (PMID:20471975). Page 2 of 3 v.20.1 Updated 2/9/2021 Images Western Blot: ZBTB7A/Pokemon Antibody [NB100-761] - Whole cell lysate (50 ug) from TCMK-1 and NIH 3T3 cells prepared using NETN lysis buffer. Antibody: Affinity purified rabbit anti-ZBTB7/FBI-1 antibody used for WB at 0.1 ug/ml. Detection: Chemiluminescence with an exposure time of 3 minutes. -

Antibodies to Transcription Factors and Regulators Over 2,000 Antibodies Targeting More Than 900 Proteins
Really Good Antibodies Antibodies to Transcription Factors and Regulators Over 2,000 Antibodies Targeting More Than 900 Proteins bethyl.com Bethyl Laboratories, Inc. has been dedicated to supporting scientific discovery through its qualified antibody products and custom antibody services since its founding in 1972. Every antibody that Bethyl sells has been manufactured to exacting standards at its sole location in Montgomery, Texas, and has been validated in-house by Bethyl’s team of scientists. Antibodies are tested across a range of applications including western blot, immunoprecipitation, immunohistochemistry, immunocytochemistry, ChIP, proximity ligation assay and ELISA. Currently, Bethyl’s portfolio consists of over 7,150 catalog products; offering close to 5,700 primary antibodies targeting over 2,700 proteins and 1,450 secondary antibodies raised against immunoglobulins from over 25 species. With over 40 years of experience, Bethyl is also a leading provider of custom antibody production services. Bethyl offers complete packages from initial peptide synthesis to affinity purification of your specific antibody from an antigen-specific immunosorbent. Because of its rigorous validation process and high standards, Bethyl does not sell every antibody it makes. Bethyl serves to advance science by concentrating its resources on developing qualified antibodies, including many to emerging and underserved protein targets. Below is a sampling of some of Bethyl’s more than 2,000 antibodies targeting over 900 transcription factors and regulators. Please visit www.bethyl.com/category/pca_research_TRANSCRIPTION for a complete listing. Detection of human phospho-RNA polymerase II (S5) in prostate carcinoma by Transcriptional Research Antibodies immunohistochemistry. Antibody: Rabbit anti- Phospho-RNA Polymerase II (S5), Cat# IHC-00387. -

Role of the Proto-Oncogene Pokemon in Cellular Transformation and ARF Repression
articles Role of the proto-oncogene Pokemon in cellular transformation and ARF repression Takahiro Maeda1,2, Robin M. Hobbs1,2, Taha Merghoub1,2, Ilhem Guernah1,2, Arthur Zelent3, Carlos Cordon-Cardo2, Julie Teruya-Feldstein2 & Pier Paolo Pandolfi1,2 1Cancer Biology and Genetics Program, 2Department of Pathology, Memorial Sloan-Kettering Cancer Center, Sloan-Kettering Institute, 1275 York Avenue, New York, New York 10021, USA 3Leukemia Research Fund Center at the Institute of Cancer Research, Chester Beatty Laboratories, Fulham Road, London SW3 6JB, UK ........................................................................................................................................................................................................................... Aberrant transcriptional repression through chromatin remodelling and histone deacetylation has been postulated to represent a driving force underlying tumorigenesis because histone deacetylase inhibitors have been found to be effective in cancer treatment. However, the molecular mechanisms by which transcriptional derepression would be linked to tumour suppression are poorly understood. Here we identify the transcriptional repressor Pokemon (encoded by the Zbtb7 gene) as a critical factor in oncogenesis. Mouse embryonic fibroblasts lacking Zbtb7 are completely refractory to oncogene-mediated cellular transformation. Conversely, Pokemon overexpression leads to overt oncogenic transformation both in vitro and in vivo in transgenic mice. Pokemon can specifically repress the transcription -

Some Bioinformatic Analyses of Human GDAP1 Gene Expression
Some bioinformatic analyses of human GDAP1 gene expression Khloud Mubarak Algothmi Thesis submitted, in fulfilment of the requirements for the degree of Doctor of Philosophy in Applied science. University of Canberra July 2015 Abstract Charcot- Marie-Tooth (CMT) represents a group of genetic disorders, which cause damage in the peripheral nervous system. It was identified and described in 1886 by Jean-Martin Charcot, Pierre Marie and Howard Henry Tooth. It is the most common inherited disorder of the peripheral nervous system, and affects approximately 1 in every 2,500 people. A severe form of CMT has been linked to mutations in the coding region of Ganglioside-induced Differentiation Associated Protein (GDAP1), a member of the glutathione transferase (GST) family which is located in the outer membrane of mitochondria. GDAP1 mutations cause axonal, demyelinating and intermediate forms of CMT. In some cases the same mutation can cause different CMT phenotypes. The overall hypothesis for this thesis was, that changes in the expression in GDAP1 may lead to these phenotypic differences. The methodology used to investigate the expression of human GDAP1 gene was a bioinformatic approach. The results demonstrated that in normal healthy tissues, GDAP1 had ubiquitous expression, particularly in neural and endocrine tissues. This pattern of expression was different to the expression of mouse GDAP1, where expression was predominantly in nervous tissues. GDAP1 has mainly been studied in the context of peripheral neuropathies, based on its genetic linkage with CMT disease. In this study, the expression of GDAP1 was shown to be altered in some other diseases, such as brain cancers.