© Ferrata Storti Foundation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Order Estimation Based Approach to Identify Response Genes
AN ORDER ESTIMATION BASED APPROACH TO IDENTIFY RESPONSE GENES FOR MICRO ARRAY TIME COURSE DATA A Thesis Presented to The Faculty of Graduate Studies of The University of Guelph by ZHIHENG LU In partial fulfilment of requirements for the degree of Doctor of Philosophy September, 2008 © Zhiheng Lu, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-47605-5 Our file Notre reference ISBN: 978-0-494-47605-5 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -
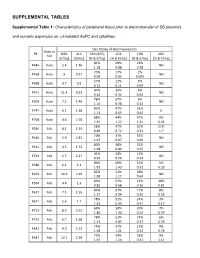
Supplemental Tables
SUPPLEMENTAL TABLES Supplemental Table 1: Characteristics of peripheral blood prior to electrotransfer of SB plasmids and numeric expansion on γ-irradiated AaPC and cytokines Day 0 (Day of electroporation) Auto or P# WBC ALC CD3 (ATC) CD4 CD8 ABC Allo [K/mL] [K/mL] [% & K/mL] [% & K/mL] [% & K/mL] [% & K/mL] 81% 60% 19% P446 Auto 5.4 1.46 ND 1.18 0.88 0.28 23% 17% 2% P458 Auto 9 0.27 ND 0.06 0.05 0.005 17% 12% 5% P468 Auto 4.7 0.9 ND 0.15 0.11 0.04 47% 35% 5% P471 Auto 11.4 0.93 ND 0.44 0.32 0.04 78% 67% 9% P509 Auto 7.2 1.46 ND 1.14 0.98 0.13 82% 47% 32% P747 Auto 6.1 1.38 0 1.13 0.65 0.44 88% 44% 47% 0% P708 Auto 4.6 2.78 2.45 1.22 1.31 0.05 58% 47% 20% 21% P396 Allo 8.1 1.54 0.89 0.72 0.31 1.7 70% 31% 32% P410 Allo 5.9 2.81 ND 1.97 0.87 0.90 80% 48% 32% P411 Allo 4.5 1.72 ND 1.38 0.83 0.55 41% 24% 15% P513 Allo 6.7 2.27 ND 0.93 0.54 0.34 86% 68% 15% 5% P580 Allo 6.1 2.1 1.81 1.43 0.32 0.29 82% 51% 28% P459 Allo 10.6 2.29 ND 1.88 1.17 0.64 64% 52% 12% 18% P564 Allo 4.4 1.3 0.83 0.68 0.16 0.81 82% 61% 17% 8% P617 Allo 7.1 1.55 1.27 0.94 0.26 0.58 78% 53% 24% 3% P671 Allo 5.4 1.7 1.33 0.90 0.41 0.17 69% 50% 20% 7% P723 Allo 8.6 2.61 1.80 1.30 0.52 0.59 78% 52% 22% 6% P732 Allo 6.7 1.68 1.31 0.87 0.37 0.39 74% 57% 15% 9% P641 Allo 9.0 2.13 1.58 1.21 0.32 0.78 73% 54% 18% 9% P647 Allo 12.1 2.29 1.67 1.24 0.41 1.11 75% 59% 11% P716 Allo 3.0 1.83 0 1.37 1.08 0.20 87% 73% 12% 1% P718 Allo 6.2 1.99 1.73 1.45 0.24 0.07 83% 69% 13% 6% P771 Allo 13.1 3.23 2.68 2.23 0.42 0.79 73% 42% 27% 6% P783 Allo 8.1 1.54 1.12 0.65 0.42 0.52 WBC = white blood -

Subcloning, Expression, and Enzymatic Study of PRMT5
Georgia State University ScholarWorks @ Georgia State University Biology Theses Department of Biology Summer 7-12-2010 Subcloning, Expression, and Enzymatic Study of PRMT5 Ran Guo Georgia State University Follow this and additional works at: https://scholarworks.gsu.edu/biology_theses Part of the Biology Commons Recommended Citation Guo, Ran, "Subcloning, Expression, and Enzymatic Study of PRMT5." Thesis, Georgia State University, 2010. https://scholarworks.gsu.edu/biology_theses/26 This Thesis is brought to you for free and open access by the Department of Biology at ScholarWorks @ Georgia State University. It has been accepted for inclusion in Biology Theses by an authorized administrator of ScholarWorks @ Georgia State University. For more information, please contact [email protected]. SUBCLONING, EXPRESSION, AND ENZYMATIC STUDY OF PRMT5 by RAN GUO Under the Direction of Yujun George Zheng ABSTRACT Protein arginine methyltransferases (PRMTs) mediate the transfer of methyl groups to arginine residues in histone and non-histone proteins. PRMT5 is an important member of PRMTs which symmetrically dimethylates arginine 8 in histone H3 (H3R8) and arginine 3 in histone H4 (H4R3). PRMT5 was reported to inhibit some tumor suppressors in leukemia and lymphoma cells and regulate p53 gene, through affecting the promoter of p53. Through methylation of H4R3, PRMT5 can recruit DNA-methyltransferase 3A (DNMT3A) which regulates gene transcription. All the above suggest that PRMT5 has an important function of suppressing cell apoptosis and is a potential anticancer target. Currently, the enzymatic activities of PRMT5 are not clearly understood. In our study, we improved the protein expression methodology and greatly enhanced the yield and quality of the recombinant PRMT5. -

P53 — 30 Yreviewsears On
FOCUS ON P53 — 30 YREVIEWSEARS ON p53 — a Jack of all trades but master of none Melissa R. Junttila and Gerard I. Evan Abstract | Cancers are rare because their evolution is actively restrained by a range of tumour suppressors. Of these p53 seems unusually crucial as either it or its attendant upstream or downstream pathways are inactivated in virtually all cancers. p53 is an evolutionarily ancient coordinator of metazoan stress responses. Its role in tumour suppression is likely to be a relatively recent adaptation, which is only necessary when large, long-lived organisms acquired the sufficient size and somatic regenerative capacity to necessitate specific mechanisms to reign in rogue proliferating cells. However, such evolutionary reappropriation of this venerable transcription factor entails compromises that restrict its efficacy as a tumour suppressor. Cnidarians–bilaterians Cancer is a genetic pathology that arises in the adult tissues questions are important for several reasons. Knowing Cinidarians comprise an animal of long-lived organisms, such as vertebrates, whose tis- how p53 discriminates between normal and tumour cells phylum of ~9,000 radially sues retain a substantial regenerative capacity throughout might point to attributes of cancer cells that qualitatively symmetrical, mostly marine life and, consequently, whose somatic cells accumulate distinguish them from normal somatic cells and could organisms. Most other animals are bilaterally symmetrical mutations. Occasionally, such mutations corrupt the be used as tumour-specific targets. Knowing what sig- and are classed as bilateria. regulatory mechanisms that suppress untoward somatic nals drive selection for loss of p53 function in cancers The cnidarians and bilaterians cell proliferation, survival and migration, resulting in the would help us understand what constrains and dictates last shared a common ancestor progressive outgrowth of a somatic clone. -

A Flexible Microfluidic System for Single-Cell Transcriptome Profiling
www.nature.com/scientificreports OPEN A fexible microfuidic system for single‑cell transcriptome profling elucidates phased transcriptional regulators of cell cycle Karen Davey1,7, Daniel Wong2,7, Filip Konopacki2, Eugene Kwa1, Tony Ly3, Heike Fiegler2 & Christopher R. Sibley 1,4,5,6* Single cell transcriptome profling has emerged as a breakthrough technology for the high‑resolution understanding of complex cellular systems. Here we report a fexible, cost‑efective and user‑ friendly droplet‑based microfuidics system, called the Nadia Instrument, that can allow 3′ mRNA capture of ~ 50,000 single cells or individual nuclei in a single run. The precise pressure‑based system demonstrates highly reproducible droplet size, low doublet rates and high mRNA capture efciencies that compare favorably in the feld. Moreover, when combined with the Nadia Innovate, the system can be transformed into an adaptable setup that enables use of diferent bufers and barcoded bead confgurations to facilitate diverse applications. Finally, by 3′ mRNA profling asynchronous human and mouse cells at diferent phases of the cell cycle, we demonstrate the system’s ability to readily distinguish distinct cell populations and infer underlying transcriptional regulatory networks. Notably this provided supportive evidence for multiple transcription factors that had little or no known link to the cell cycle (e.g. DRAP1, ZKSCAN1 and CEBPZ). In summary, the Nadia platform represents a promising and fexible technology for future transcriptomic studies, and other related applications, at cell resolution. Single cell transcriptome profling has recently emerged as a breakthrough technology for understanding how cellular heterogeneity contributes to complex biological systems. Indeed, cultured cells, microorganisms, biopsies, blood and other tissues can be rapidly profled for quantifcation of gene expression at cell resolution. -

The Function and Evolution of C2H2 Zinc Finger Proteins and Transposons
The function and evolution of C2H2 zinc finger proteins and transposons by Laura Francesca Campitelli A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Molecular Genetics University of Toronto © Copyright by Laura Francesca Campitelli 2020 The function and evolution of C2H2 zinc finger proteins and transposons Laura Francesca Campitelli Doctor of Philosophy Department of Molecular Genetics University of Toronto 2020 Abstract Transcription factors (TFs) confer specificity to transcriptional regulation by binding specific DNA sequences and ultimately affecting the ability of RNA polymerase to transcribe a locus. The C2H2 zinc finger proteins (C2H2 ZFPs) are a TF class with the unique ability to diversify their DNA-binding specificities in a short evolutionary time. C2H2 ZFPs comprise the largest class of TFs in Mammalian genomes, including nearly half of all Human TFs (747/1,639). Positive selection on the DNA-binding specificities of C2H2 ZFPs is explained by an evolutionary arms race with endogenous retroelements (EREs; copy-and-paste transposable elements), where the C2H2 ZFPs containing a KRAB repressor domain (KZFPs; 344/747 Human C2H2 ZFPs) are thought to diversify to bind new EREs and repress deleterious transposition events. However, evidence of the gain and loss of KZFP binding sites on the ERE sequence is sparse due to poor resolution of ERE sequence evolution, despite the recent publication of binding preferences for 242/344 Human KZFPs. The goal of my doctoral work has been to characterize the Human C2H2 ZFPs, with specific interest in their evolutionary history, functional diversity, and coevolution with LINE EREs. -

Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Integration of 198 ChIP-seq datasets reveals human cis-regulatory regions Hamid Bolouri & Larry Ruzzo Thanks to Stephen Tapscott, Steven Henikoff & Zizhen Yao Slides will be available from: http://labs.fhcrc.org/bolouri/ Email [email protected] for manuscript (Bolouri & Ruzzo, submitted) Kleinjan & van Heyningen, Am. J. Hum. Genet., 2005, (76)8–32 Epstein, Briefings in Func. Genom. & Protemoics, 2009, 8(4)310-16 Regulation of SPi1 (Sfpi1, PU.1 protein) expression – part 1 miR155*, miR569# ~750nt promoter ~250nt promoter The antisense RNA • causes translational stalling • has its own promoter • requires distal SPI1 enhancer • is transcribed with/without SPI1. # Hikami et al, Arthritis & Rheumatism, 2011, 63(3):755–763 * Vigorito et al, 2007, Immunity 27, 847–859 Ebralidze et al, Genes & Development, 2008, 22: 2085-2092. Regulation of SPi1 expression – part 2 (mouse coordinates) Bidirectional ncRNA transcription proportional to PU.1 expression PU.1/ELF1/FLI1/GLI1 GATA1 GATA1 Sox4/TCF/LEF PU.1 RUNX1 SP1 RUNX1 RUNX1 SP1 ELF1 NF-kB SATB1 IKAROS PU.1 cJun/CEBP OCT1 cJun/CEBP 500b 500b 500b 500b 500b 750b 500b -18Kb -14Kb -12Kb -10Kb -9Kb Chou et al, Blood, 2009, 114: 983-994 Hoogenkamp et al, Molecular & Cellular Biology, 2007, 27(21):7425-7438 Zarnegar & Rothenberg, 2010, Mol. & cell Biol. 4922-4939 An NF-kB binding-site variant in the SPI1 URE reduces PU.1 expression & is GGGCCTCCCC correlated with AML GGGTCTTCCC Bonadies et al, Oncogene, 2009, 29(7):1062-72. SATB1 binding site A distal single nucleotide polymorphism alters long- range regulation of the PU.1 gene in acute myeloid leukemia Steidl et al, J Clin Invest. -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Methodology article Open Access "NeuroStem Chip": a novel highly specialized tool to study neural differentiation pathways in human stem cells Sergey V Anisimov*, Nicolaj S Christophersen, Ana S Correia, Jia-Yi Li and Patrik Brundin Address: Neuronal Survival Unit, Wallenberg Neuroscience Center, Lund University, 221 84 Lund, Sweden Email: Sergey V Anisimov* - [email protected]; Nicolaj S Christophersen - [email protected]; Ana S Correia - [email protected]; Jia-Yi Li - [email protected]; Patrik Brundin - [email protected] * Corresponding author Published: 8 February 2007 Received: 21 September 2006 Accepted: 8 February 2007 BMC Genomics 2007, 8:46 doi:10.1186/1471-2164-8-46 This article is available from: http://www.biomedcentral.com/1471-2164/8/46 © 2007 Anisimov et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Human stem cells are viewed as a possible source of neurons for a cell-based therapy of neurodegenerative disorders, such as Parkinson's disease. Several protocols that generate different types of neurons from human stem cells (hSCs) have been developed. Nevertheless, the cellular mechanisms that underlie the development of neurons in vitro as they are subjected to the specific differentiation protocols are often poorly understood. Results: We have designed a focused DNA (oligonucleotide-based) large-scale microarray platform (named "NeuroStem Chip") and used it to study gene expression patterns in hSCs as they differentiate into neurons. -

Prostate News
Prostate Cancer and Prostatic Diseases (2005) 8, 2–6 & 2005 Nature Publishing Group All rights reserved 1365-7852/05 $30.00 www.nature.com/pcan Research Highlights Prostate News Prostate Cancer and Prostatic Diseases (2005) 8, 2–6. doi:10.1038/sj.pcan.4500793 Pokemon’s central role in cancer progression of prostate cancer. Epigenetic changes are reversible, which make them attractive targets for cancer development treatment with modulators that demethylate DNA and Researchers at New York’s Memorial Sloan-Kettering inhibit histone deacetylases, leading to reactivation of Cancer Center report novel findings in Nature regarding silenced genes. DNA methylation and histone modifica- a new cellular oncogene implicated in the development tions are important epigenetic mechanisms of gene of human cancers, including prostate neoplasia. Poke- regulation, and both play essential roles in tumor mon, a pop-culture inspired acronym for ‘POK erythroid initiation and progression. In prostate cancer, aberrant myeloid ontogenic factor’, is a POK protein family epigenetic events such as DNA hypo- and hypermethy- member encoded by the ZbTb7 gene and characterized lation and altered histone acetylation have been by its critical transcription function in cellular differ- observed to affect a large number of genes. However, entiation. Takahiro Maeda, Pier Paolo Pandolfi and no plenitude is without peril: although the number of colleagues show that Pokemon acts as a genuine proto- genes that undergo aberrant epigenetic inactivation oncogene when overexpressed by inhibiting the expres- associated with prostate cancer seems to be growing, sion of tumor suppressors, such as ARF. very few of these genes have yielded promising results To establish this novel finding, the authors conducted as potential tumor biomarkers for early diagnosis and a complete battery of genetic, clinical, and pathological risk assessment. -

Supplementary Material Computational Prediction of SARS
Supplementary_Material Computational prediction of SARS-CoV-2 encoded miRNAs and their putative host targets Sheet_1 List of potential stem-loop structures in SARS-CoV-2 genome as predicted by VMir. Rank Name Start Apex Size Score Window Count (Absolute) Direct Orientation 1 MD13 2801 2864 125 243.8 61 2 MD62 11234 11286 101 211.4 49 4 MD136 27666 27721 104 205.6 119 5 MD108 21131 21184 110 204.7 210 9 MD132 26743 26801 119 188.9 252 19 MD56 9797 9858 128 179.1 59 26 MD139 28196 28233 72 170.4 133 28 MD16 2934 2974 76 169.9 71 43 MD103 20002 20042 80 159.3 403 46 MD6 1489 1531 86 156.7 171 51 MD17 2981 3047 131 152.8 38 87 MD4 651 692 75 140.3 46 95 MD7 1810 1872 121 137.4 58 116 MD140 28217 28252 72 133.8 62 122 MD55 9712 9758 96 132.5 49 135 MD70 13171 13219 93 130.2 131 164 MD95 18782 18820 79 124.7 184 173 MD121 24086 24135 99 123.1 45 176 MD96 19046 19086 75 123.1 179 196 MD19 3197 3236 76 120.4 49 200 MD86 17048 17083 73 119.8 428 223 MD75 14534 14600 137 117 51 228 MD50 8824 8870 94 115.8 79 234 MD129 25598 25642 89 115.6 354 Reverse Orientation 6 MR61 19088 19132 88 197.8 271 10 MR72 23563 23636 148 188.8 286 11 MR11 3775 3844 136 185.1 116 12 MR94 29532 29582 94 184.6 271 15 MR43 14973 15028 109 183.9 226 27 MR14 4160 4206 89 170 241 34 MR35 11734 11792 111 164.2 37 52 MR5 1603 1652 89 152.7 118 53 MR57 18089 18132 101 152.7 139 94 MR8 2804 2864 122 137.4 38 107 MR58 18474 18508 72 134.9 237 117 MR16 4506 4540 72 133.8 311 120 MR34 10010 10048 82 132.7 245 133 MR7 2534 2578 90 130.4 75 146 MR79 24766 24808 75 127.9 59 150 MR65 21528 21576 99 127.4 83 180 MR60 19016 19049 70 122.5 72 187 MR51 16450 16482 75 121 363 190 MR80 25687 25734 96 120.6 75 198 MR64 21507 21544 70 120.3 35 206 MR41 14500 14542 84 119.2 94 218 MR84 26840 26894 108 117.6 94 Sheet_2 List of stable stem-loop structures based on MFE. -

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: • This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. • A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. • This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. • The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. • When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. Regulation and Function of miR-199-3p in Murine and Human Cytomegalovirus Infections Nouf N. M. Laqtom s0898296 A thesis submitted in fulfilment of requirements for the degree of Doctor of Philosophy Division of Pathway Medicine, School of Biomedical Sciences The University of Edinburgh May 2013 Supervisors: Dr. Amy Buck and Dr. Bernadette Dutia i Declaration of Authorship I hereby declare that this thesis is of my own composition, and that it contains no material previously submitted for the award of any other degree. The work reported in this thesis has been executed by myself, except where due acknowledgement is made in the text. Nouf N. M. Laqtom ii Abstract Human Cytomegalovirus (HCMV), the prototypic β-herpesvirus, is the most common cause of congenital infections as well as morbidity and mortality in immunocompromised patients. -

Establishment of Mouse Primed Stem Cells by Combination of Activin and LIF Signaling
fcell-09-713503 July 31, 2021 Time: 12:43 # 1 ORIGINAL RESEARCH published: 05 August 2021 doi: 10.3389/fcell.2021.713503 Establishment of Mouse Primed Stem Cells by Combination of Activin and LIF Signaling Mengyi Wei1,2†, Yanglin Chen1,2,3†, Chaoyue Zhao1,2, Li Zheng1,2, Baojiang Wu1,2, Chen Chen1,2, Xihe Li1,2,4 and Siqin Bao1,2* 1 State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock, Inner Mongolia University, Hohhot, China, 2 Institute of Animal Genetic Research of Mongolia Plateau, College of Life Sciences, Inner Mongolia University, Hohhot, China, 3 School of Basic Medical Sciences, Southern Medical University, Guangzhou, China, 4 Inner Mongolia Saikexing Institute of Breeding and Reproductive Biotechnology in Domestic Animal, Hohhot, China In mice, embryonic stem cells (ESCs) and epiblast stem cells (EpiSCs) are established from pre- and post-implantation embryos and represent the naive and primed state, respectively. Herein we used mouse leukemia inhibitory factor (LIF), which supports Edited by: Wei Jiang, ESCs self-renewal and Activin A (Act A), which is the main factor in maintaining EpiSCs Wuhan University, China in post-implantation epiblast cultures, to derive a primed stem cell line named ALSCs. Reviewed by: Like EpiSCs, ALSCs express key pluripotent genes Oct4, Sox2, and Nanog; one X Tetsuya S. Tanaka, Elixirgen Scientific, Inc., United States chromosome was inactivated; and the cells failed to contribute to chimera formation Fabiana Passaro, in vivo. Notably, compared to EpiSCs, ALSCs efficiently reversed to ESCs (rESCs) University of Naples Federico II, Italy on activation of Wnt signaling. Moreover, we also discovered that culturing EpiSCs in *Correspondence: AL medium for several passages favored Wnt signaling-driven naive pluripotency.