An Order Estimation Based Approach to Identify Response Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Characterization of PRE-1 Reveals a Hidden Evolutionary Relationship Between Suidae and Primates
bioRxiv preprint doi: https://doi.org/10.1101/025791; this version posted August 31, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genome-wide characterization of PRE-1 reveals a hidden evolutionary relationship between suidae and primates Hao Yu1,, Qingyan Wu1, Jing Zhag1, Ying zhang1, Chao Lu1, Yunyun Cheng1, Zhihui Zhao1, Andreas Windemuth3, Di Liu2,, Linlin Hao1 1 College of Animal Science, Jilin University, Changchun 130062, China 2 Heilongjiang Academy of Agricultural Sciences, Harbin 150086, China 3 Abcam, Firefly BioWorks Inc, United States Corresponding author: Linlin Hao ([email protected]); Di liu ([email protected]); Andreas Windemuth ([email protected]) Abstract We identified and characterized a free PRE-1 element inserted into the promoter region of the porcine IGFBP7 gene whose integration mechanisms into the genome, including copy number, distribution preferences, capacity to exonize and phyloclustering pattern are similar to that of the primate Alu element. 98% of these PRE-1 elements also contain two conserved internal AluI restriction enzyme recognition sites, and the RNA structure of PRE1 can be folded into a two arms model like the Alu RNA structure. It is more surprising that the length of the PRE-1 fragments is nearly the same in 20 chromosomes and positively correlated to its fracture site frequency. All of these fracture sites are close to the mutation hot spots of PRE-1 families, and most of these hot spots are located in the non-complementary fragile regions of the PRE-1 RNA structure. -

The Title of the Article
Mechanism-Anchored Profiling Derived from Epigenetic Networks Predicts Outcome in Acute Lymphoblastic Leukemia Xinan Yang, PhD1, Yong Huang, MD1, James L Chen, MD1, Jianming Xie, MSc2, Xiao Sun, PhD2, Yves A Lussier, MD1,3,4§ 1Center for Biomedical Informatics and Section of Genetic Medicine, Department of Medicine, The University of Chicago, Chicago, IL 60637 USA 2State Key Laboratory of Bioelectronics, Southeast University, 210096 Nanjing, P.R.China 3The University of Chicago Cancer Research Center, and The Ludwig Center for Metastasis Research, The University of Chicago, Chicago, IL 60637 USA 4The Institute for Genomics and Systems Biology, and the Computational Institute, The University of Chicago, Chicago, IL 60637 USA §Corresponding author Email addresses: XY: [email protected] YH: [email protected] JC: [email protected] JX: [email protected] XS: [email protected] YL: [email protected] - 1 - Abstract Background Current outcome predictors based on “molecular profiling” rely on gene lists selected without consideration for their molecular mechanisms. This study was designed to demonstrate that we could learn about genes related to a specific mechanism and further use this knowledge to predict outcome in patients – a paradigm shift towards accurate “mechanism-anchored profiling”. We propose a novel algorithm, PGnet, which predicts a tripartite mechanism-anchored network associated to epigenetic regulation consisting of phenotypes, genes and mechanisms. Genes termed as GEMs in this network meet all of the following criteria: (i) they are co-expressed with genes known to be involved in the biological mechanism of interest, (ii) they are also differentially expressed between distinct phenotypes relevant to the study, and (iii) as a biomodule, genes correlate with both the mechanism and the phenotype. -
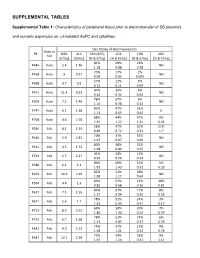
Supplemental Tables
SUPPLEMENTAL TABLES Supplemental Table 1: Characteristics of peripheral blood prior to electrotransfer of SB plasmids and numeric expansion on γ-irradiated AaPC and cytokines Day 0 (Day of electroporation) Auto or P# WBC ALC CD3 (ATC) CD4 CD8 ABC Allo [K/mL] [K/mL] [% & K/mL] [% & K/mL] [% & K/mL] [% & K/mL] 81% 60% 19% P446 Auto 5.4 1.46 ND 1.18 0.88 0.28 23% 17% 2% P458 Auto 9 0.27 ND 0.06 0.05 0.005 17% 12% 5% P468 Auto 4.7 0.9 ND 0.15 0.11 0.04 47% 35% 5% P471 Auto 11.4 0.93 ND 0.44 0.32 0.04 78% 67% 9% P509 Auto 7.2 1.46 ND 1.14 0.98 0.13 82% 47% 32% P747 Auto 6.1 1.38 0 1.13 0.65 0.44 88% 44% 47% 0% P708 Auto 4.6 2.78 2.45 1.22 1.31 0.05 58% 47% 20% 21% P396 Allo 8.1 1.54 0.89 0.72 0.31 1.7 70% 31% 32% P410 Allo 5.9 2.81 ND 1.97 0.87 0.90 80% 48% 32% P411 Allo 4.5 1.72 ND 1.38 0.83 0.55 41% 24% 15% P513 Allo 6.7 2.27 ND 0.93 0.54 0.34 86% 68% 15% 5% P580 Allo 6.1 2.1 1.81 1.43 0.32 0.29 82% 51% 28% P459 Allo 10.6 2.29 ND 1.88 1.17 0.64 64% 52% 12% 18% P564 Allo 4.4 1.3 0.83 0.68 0.16 0.81 82% 61% 17% 8% P617 Allo 7.1 1.55 1.27 0.94 0.26 0.58 78% 53% 24% 3% P671 Allo 5.4 1.7 1.33 0.90 0.41 0.17 69% 50% 20% 7% P723 Allo 8.6 2.61 1.80 1.30 0.52 0.59 78% 52% 22% 6% P732 Allo 6.7 1.68 1.31 0.87 0.37 0.39 74% 57% 15% 9% P641 Allo 9.0 2.13 1.58 1.21 0.32 0.78 73% 54% 18% 9% P647 Allo 12.1 2.29 1.67 1.24 0.41 1.11 75% 59% 11% P716 Allo 3.0 1.83 0 1.37 1.08 0.20 87% 73% 12% 1% P718 Allo 6.2 1.99 1.73 1.45 0.24 0.07 83% 69% 13% 6% P771 Allo 13.1 3.23 2.68 2.23 0.42 0.79 73% 42% 27% 6% P783 Allo 8.1 1.54 1.12 0.65 0.42 0.52 WBC = white blood -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Sequence Analysis of Familial Neurodevelopmental Disorders
SEQUENCE ANALYSIS OF FAMILIAL NEURODEVELOPMENTAL DISORDERS by Joseph Mark Tilghman A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland December 2020 © 2020 Joseph Tilghman All Rights Reserved Abstract: In the practice of human genetics, there is a gulf between the study of Mendelian and complex inheritance. When diagnosis of families affected by presumed monogenic syndromes is undertaken by genomic sequencing, these families are typically considered to have been solved only when a single gene or variant showing apparently Mendelian inheritance is discovered. However, about half of such families remain unexplained through this approach. On the other hand, common regulatory variants conferring low risk of disease still predominate our understanding of individual disease risk in complex disorders, despite rapidly increasing access to rare variant genotypes through sequencing. This dissertation utilizes primarily exome sequencing across several developmental disorders (having different levels of genetic complexity) to investigate how to best use an individual’s combination of rare and common variants to explain genetic risk, phenotypic heterogeneity, and the molecular bases of disorders ranging from those presumed to be monogenic to those known to be highly complex. The study described in Chapter 2 addresses putatively monogenic syndromes, where we used exome sequencing of four probands having syndromic neurodevelopmental disorders from an Israeli-Arab founder population to diagnose recessive and dominant disorders, highlighting the need to consider diverse modes of inheritance and phenotypic heterogeneity. In the study described in Chapter 3, we address the case of a relatively tractable multifactorial disorder, Hirschsprung disease. -

Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer
GENES, CHROMOSOMES & CANCER 47:1049–1066 (2008) Differential Patterns of Allelic Loss in Estrogen Receptor-Positive Infiltrating Lobular and Ductal Breast Cancer L. W. M. Loo,1 C. Ton,1,2 Y.-W. Wang,2 D. I. Grove,2 H. Bouzek,1 N. Vartanian,1 M.-G. Lin,1 X. Yuan,1 T. L. Lawton,3 J. R. Daling,2 K. E. Malone,2 C. I. Li,2 L. Hsu,2 and P.L. Porter1,2,3* 1Division of Human Biology,Fred Hutchinson Cancer Research Center,Seattle,WA 2Division of Public Health Sciences,Fred Hutchinson Cancer Research Center,Seattle,WA 3Departmentof Pathology,Universityof Washington,Seattle,WA The two main histological types of infiltrating breast cancer, lobular (ILC) and the more common ductal (IDC) carcinoma are morphologically and clinically distinct. To assess the molecular alterations associated with these breast cancer subtypes, we conducted a whole-genome study of 166 archival estrogen receptor (ER)-positive tumors (89 IDC and 77 ILC) using the Affy- metrix GeneChip® Mapping 10K Array to identify sites of loss of heterozygosity (LOH) that either distinguished, or were shared by, the two phenotypes. We found single nucleotide polymorphisms (SNPs) of high-frequency LOH (>50%) common to both ILC and IDC tumors predominately in 11q, 16q, and 17p. Overall, IDC had a slightly higher frequency of LOH events across the genome than ILC (fractional allelic loss 5 0.186 and 0.156). By comparing the average frequency of LOH by chro- mosomal arm, we found IDC tumors with significantly (P < 0.05) higher frequency of LOH on 3p, 5q, 8p, 9p, 20p, and 20q than ILC tumors. -

P53 — 30 Yreviewsears On
FOCUS ON P53 — 30 YREVIEWSEARS ON p53 — a Jack of all trades but master of none Melissa R. Junttila and Gerard I. Evan Abstract | Cancers are rare because their evolution is actively restrained by a range of tumour suppressors. Of these p53 seems unusually crucial as either it or its attendant upstream or downstream pathways are inactivated in virtually all cancers. p53 is an evolutionarily ancient coordinator of metazoan stress responses. Its role in tumour suppression is likely to be a relatively recent adaptation, which is only necessary when large, long-lived organisms acquired the sufficient size and somatic regenerative capacity to necessitate specific mechanisms to reign in rogue proliferating cells. However, such evolutionary reappropriation of this venerable transcription factor entails compromises that restrict its efficacy as a tumour suppressor. Cnidarians–bilaterians Cancer is a genetic pathology that arises in the adult tissues questions are important for several reasons. Knowing Cinidarians comprise an animal of long-lived organisms, such as vertebrates, whose tis- how p53 discriminates between normal and tumour cells phylum of ~9,000 radially sues retain a substantial regenerative capacity throughout might point to attributes of cancer cells that qualitatively symmetrical, mostly marine life and, consequently, whose somatic cells accumulate distinguish them from normal somatic cells and could organisms. Most other animals are bilaterally symmetrical mutations. Occasionally, such mutations corrupt the be used as tumour-specific targets. Knowing what sig- and are classed as bilateria. regulatory mechanisms that suppress untoward somatic nals drive selection for loss of p53 function in cancers The cnidarians and bilaterians cell proliferation, survival and migration, resulting in the would help us understand what constrains and dictates last shared a common ancestor progressive outgrowth of a somatic clone. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Material
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Page 1 / 45 SUPPLEMENTARY MATERIAL Appendix A1: Neuropsychological protocol. Appendix A2: Description of the four cases at the transitional stage. Table A1: Clinical status and center proportion in each batch. Table A2: Complete output from EdgeR. Table A3: List of the putative target genes. Table A4: Complete output from DIANA-miRPath v.3. Table A5: Comparison of studies investigating miRNAs from brain samples. Figure A1: Stratified nested cross-validation. Figure A2: Expression heatmap of miRNA signature. Figure A3: Bootstrapped ROC AUC scores. Figure A4: ROC AUC scores with 100 different fold splits. Figure A5: Presymptomatic subjects probability scores. Figure A6: Heatmap of the level of enrichment in KEGG pathways. Kmetzsch V, et al. J Neurol Neurosurg Psychiatry 2021; 92:485–493. doi: 10.1136/jnnp-2020-324647 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Neurol Neurosurg Psychiatry Appendix A1. Neuropsychological protocol The PREV-DEMALS cognitive evaluation included standardized neuropsychological tests to investigate all cognitive domains, and in particular frontal lobe functions. The scores were provided previously (Bertrand et al., 2018). Briefly, global cognitive efficiency was evaluated by means of Mini-Mental State Examination (MMSE) and Mattis Dementia Rating Scale (MDRS). Frontal executive functions were assessed with Frontal Assessment Battery (FAB), forward and backward digit spans, Trail Making Test part A and B (TMT-A and TMT-B), Wisconsin Card Sorting Test (WCST), and Symbol-Digit Modalities test. -

Apoptotic Genes As Potential Markers of Metastatic Phenotype in Human Osteosarcoma Cell Lines
17-31 10/12/07 14:53 Page 17 INTERNATIONAL JOURNAL OF ONCOLOGY 32: 17-31, 2008 17 Apoptotic genes as potential markers of metastatic phenotype in human osteosarcoma cell lines CINZIA ZUCCHINI1, ANNA ROCCHI2, MARIA CRISTINA MANARA2, PAOLA DE SANCTIS1, CRISTINA CAPANNI3, MICHELE BIANCHINI1, PAOLO CARINCI1, KATIA SCOTLANDI2 and LUISA VALVASSORI1 1Dipartimento di Istologia, Embriologia e Biologia Applicata, Università di Bologna, Via Belmeloro 8, 40126 Bologna; 2Laboratorio di Ricerca Oncologica, Istituti Ortopedici Rizzoli; 3IGM-CNR, Unit of Bologna, c/o Istituti Ortopedici Rizzoli, Via di Barbiano 1/10, 40136 Bologna, Italy Received May 29, 2007; Accepted July 19, 2007 Abstract. Metastasis is the most frequent cause of death among malignant primitive bone tumor, usually developing in children patients with osteosarcoma. We have previously demonstrated and adolescents, with a high tendency to metastasize (2). in independent experiments that the forced expression of Metastases in osteosarcoma patients spread through peripheral L/B/K ALP and CD99 in U-2 OS osteosarcoma cell lines blood very early and colonize primarily the lung, and later markedly reduces the metastatic ability of these cancer cells. other skeleton districts (3). Since disseminated hidden micro- This behavior makes these cell lines a useful model to assess metastases are present in 80-90% of OS patients at the time the intersection of multiple and independent gene expression of diagnosis, the identification of markers of invasiveness signatures concerning the biological problem of dissemination. and metastasis forms a target of paramount importance in With the aim to characterize a common transcriptional profile planning the treatment of osteosarcoma lesions and enhancing reflecting the essential features of metastatic behavior, we the prognosis. -

Valproic Acid Reversed Pathologic Endothelial Cell Gene Expression Profile Associated with Ischemia– Reperfusion Injury in a Swine Hemorrhagic Shock Model
From the Peripheral Vascular Surgery Society Valproic acid reversed pathologic endothelial cell gene expression profile associated with ischemia– reperfusion injury in a swine hemorrhagic shock model Marlin Wayne Causey, MD,a Shashikumar Salgar, PhD,b Niten Singh, MD,a Matthew Martin, MD,a and Jonathan D. Stallings, PhD,b Tacoma, Wash Background: Vascular endothelial cells serve as the first line of defense for end organs after ischemia and reperfusion injuries. The full etiology of this dysfunction is poorly understood, and valproic acid (VPA) has proven to be beneficial after traumatic injury. The purpose of this study was to determine the mechanism of action through which VPA exerts its beneficial effects. Methods: Sixteen Yorkshire swine underwent a standardized protocol for an ischemia–reperfusion injury through received (6 ؍ hemorrhage and a supraceliac cross-clamp with ensuing 6-hour resuscitation. The experimental swine (n Aortic .(5 ؍ and injury-control models (n (5 ؍ VPA at cross-clamp application and were compared with sham (n endothelium was harvested, and microarray analysis was performed along with a functional clustering analysis with gene transcript validation using relative quantitative polymerase chain reaction. Results: Clinical comparison of experimental swine matched for sex, weight, and length demonstrated that VPA significantly decreased resuscitative requirements, with improved hemodynamics and physiologic laboratory measure- ments. Six transcript profiles from the VPA treatment were compared with the 1536 gene transcripts (529 up and 1007 down) from sham and injury-control swine. Microarray analysis and a Database for Annotation, Visualization and Integrated Discovery functional pathway analysis approach identified biologic processes associated with pathologic vascular endothelial function, specifically through functional cluster pathways involving apoptosis/cell death and angiogenesis/vascular development, with five specific genes (THBS1, TNFRSF12A, ANGPTL4, RHOB, and RTN4) identified as members of both functional clusters.