© 2016 Ying Li ALL RIGHTS RESERVED
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Order Estimation Based Approach to Identify Response Genes
AN ORDER ESTIMATION BASED APPROACH TO IDENTIFY RESPONSE GENES FOR MICRO ARRAY TIME COURSE DATA A Thesis Presented to The Faculty of Graduate Studies of The University of Guelph by ZHIHENG LU In partial fulfilment of requirements for the degree of Doctor of Philosophy September, 2008 © Zhiheng Lu, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-47605-5 Our file Notre reference ISBN: 978-0-494-47605-5 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -
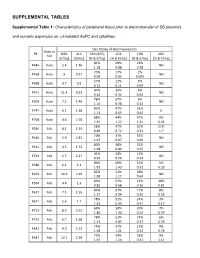
Supplemental Tables
SUPPLEMENTAL TABLES Supplemental Table 1: Characteristics of peripheral blood prior to electrotransfer of SB plasmids and numeric expansion on γ-irradiated AaPC and cytokines Day 0 (Day of electroporation) Auto or P# WBC ALC CD3 (ATC) CD4 CD8 ABC Allo [K/mL] [K/mL] [% & K/mL] [% & K/mL] [% & K/mL] [% & K/mL] 81% 60% 19% P446 Auto 5.4 1.46 ND 1.18 0.88 0.28 23% 17% 2% P458 Auto 9 0.27 ND 0.06 0.05 0.005 17% 12% 5% P468 Auto 4.7 0.9 ND 0.15 0.11 0.04 47% 35% 5% P471 Auto 11.4 0.93 ND 0.44 0.32 0.04 78% 67% 9% P509 Auto 7.2 1.46 ND 1.14 0.98 0.13 82% 47% 32% P747 Auto 6.1 1.38 0 1.13 0.65 0.44 88% 44% 47% 0% P708 Auto 4.6 2.78 2.45 1.22 1.31 0.05 58% 47% 20% 21% P396 Allo 8.1 1.54 0.89 0.72 0.31 1.7 70% 31% 32% P410 Allo 5.9 2.81 ND 1.97 0.87 0.90 80% 48% 32% P411 Allo 4.5 1.72 ND 1.38 0.83 0.55 41% 24% 15% P513 Allo 6.7 2.27 ND 0.93 0.54 0.34 86% 68% 15% 5% P580 Allo 6.1 2.1 1.81 1.43 0.32 0.29 82% 51% 28% P459 Allo 10.6 2.29 ND 1.88 1.17 0.64 64% 52% 12% 18% P564 Allo 4.4 1.3 0.83 0.68 0.16 0.81 82% 61% 17% 8% P617 Allo 7.1 1.55 1.27 0.94 0.26 0.58 78% 53% 24% 3% P671 Allo 5.4 1.7 1.33 0.90 0.41 0.17 69% 50% 20% 7% P723 Allo 8.6 2.61 1.80 1.30 0.52 0.59 78% 52% 22% 6% P732 Allo 6.7 1.68 1.31 0.87 0.37 0.39 74% 57% 15% 9% P641 Allo 9.0 2.13 1.58 1.21 0.32 0.78 73% 54% 18% 9% P647 Allo 12.1 2.29 1.67 1.24 0.41 1.11 75% 59% 11% P716 Allo 3.0 1.83 0 1.37 1.08 0.20 87% 73% 12% 1% P718 Allo 6.2 1.99 1.73 1.45 0.24 0.07 83% 69% 13% 6% P771 Allo 13.1 3.23 2.68 2.23 0.42 0.79 73% 42% 27% 6% P783 Allo 8.1 1.54 1.12 0.65 0.42 0.52 WBC = white blood -

Table S1 the Four Gene Sets Derived from Gene Expression Profiles of Escs and Differentiated Cells
Table S1 The four gene sets derived from gene expression profiles of ESCs and differentiated cells Uniform High Uniform Low ES Up ES Down EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol EntrezID GeneSymbol 269261 Rpl12 11354 Abpa 68239 Krt42 15132 Hbb-bh1 67891 Rpl4 11537 Cfd 26380 Esrrb 15126 Hba-x 55949 Eef1b2 11698 Ambn 73703 Dppa2 15111 Hand2 18148 Npm1 11730 Ang3 67374 Jam2 65255 Asb4 67427 Rps20 11731 Ang2 22702 Zfp42 17292 Mesp1 15481 Hspa8 11807 Apoa2 58865 Tdh 19737 Rgs5 100041686 LOC100041686 11814 Apoc3 26388 Ifi202b 225518 Prdm6 11983 Atpif1 11945 Atp4b 11614 Nr0b1 20378 Frzb 19241 Tmsb4x 12007 Azgp1 76815 Calcoco2 12767 Cxcr4 20116 Rps8 12044 Bcl2a1a 219132 D14Ertd668e 103889 Hoxb2 20103 Rps5 12047 Bcl2a1d 381411 Gm1967 17701 Msx1 14694 Gnb2l1 12049 Bcl2l10 20899 Stra8 23796 Aplnr 19941 Rpl26 12096 Bglap1 78625 1700061G19Rik 12627 Cfc1 12070 Ngfrap1 12097 Bglap2 21816 Tgm1 12622 Cer1 19989 Rpl7 12267 C3ar1 67405 Nts 21385 Tbx2 19896 Rpl10a 12279 C9 435337 EG435337 56720 Tdo2 20044 Rps14 12391 Cav3 545913 Zscan4d 16869 Lhx1 19175 Psmb6 12409 Cbr2 244448 Triml1 22253 Unc5c 22627 Ywhae 12477 Ctla4 69134 2200001I15Rik 14174 Fgf3 19951 Rpl32 12523 Cd84 66065 Hsd17b14 16542 Kdr 66152 1110020P15Rik 12524 Cd86 81879 Tcfcp2l1 15122 Hba-a1 66489 Rpl35 12640 Cga 17907 Mylpf 15414 Hoxb6 15519 Hsp90aa1 12642 Ch25h 26424 Nr5a2 210530 Leprel1 66483 Rpl36al 12655 Chi3l3 83560 Tex14 12338 Capn6 27370 Rps26 12796 Camp 17450 Morc1 20671 Sox17 66576 Uqcrh 12869 Cox8b 79455 Pdcl2 20613 Snai1 22154 Tubb5 12959 Cryba4 231821 Centa1 17897 -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

P53 — 30 Yreviewsears On
FOCUS ON P53 — 30 YREVIEWSEARS ON p53 — a Jack of all trades but master of none Melissa R. Junttila and Gerard I. Evan Abstract | Cancers are rare because their evolution is actively restrained by a range of tumour suppressors. Of these p53 seems unusually crucial as either it or its attendant upstream or downstream pathways are inactivated in virtually all cancers. p53 is an evolutionarily ancient coordinator of metazoan stress responses. Its role in tumour suppression is likely to be a relatively recent adaptation, which is only necessary when large, long-lived organisms acquired the sufficient size and somatic regenerative capacity to necessitate specific mechanisms to reign in rogue proliferating cells. However, such evolutionary reappropriation of this venerable transcription factor entails compromises that restrict its efficacy as a tumour suppressor. Cnidarians–bilaterians Cancer is a genetic pathology that arises in the adult tissues questions are important for several reasons. Knowing Cinidarians comprise an animal of long-lived organisms, such as vertebrates, whose tis- how p53 discriminates between normal and tumour cells phylum of ~9,000 radially sues retain a substantial regenerative capacity throughout might point to attributes of cancer cells that qualitatively symmetrical, mostly marine life and, consequently, whose somatic cells accumulate distinguish them from normal somatic cells and could organisms. Most other animals are bilaterally symmetrical mutations. Occasionally, such mutations corrupt the be used as tumour-specific targets. Knowing what sig- and are classed as bilateria. regulatory mechanisms that suppress untoward somatic nals drive selection for loss of p53 function in cancers The cnidarians and bilaterians cell proliferation, survival and migration, resulting in the would help us understand what constrains and dictates last shared a common ancestor progressive outgrowth of a somatic clone. -

Early Growth Response 1 Regulates Hematopoietic Support and Proliferation in Human Primary Bone Marrow Stromal Cells
Hematopoiesis SUPPLEMENTARY APPENDIX Early growth response 1 regulates hematopoietic support and proliferation in human primary bone marrow stromal cells Hongzhe Li, 1,2 Hooi-Ching Lim, 1,2 Dimitra Zacharaki, 1,2 Xiaojie Xian, 2,3 Keane J.G. Kenswil, 4 Sandro Bräunig, 1,2 Marc H.G.P. Raaijmakers, 4 Niels-Bjarne Woods, 2,3 Jenny Hansson, 1,2 and Stefan Scheding 1,2,5 1Division of Molecular Hematology, Department of Laboratory Medicine, Lund University, Lund, Sweden; 2Lund Stem Cell Center, Depart - ment of Laboratory Medicine, Lund University, Lund, Sweden; 3Division of Molecular Medicine and Gene Therapy, Department of Labora - tory Medicine, Lund University, Lund, Sweden; 4Department of Hematology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands and 5Department of Hematology, Skåne University Hospital Lund, Skåne, Sweden ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2019.216648 Received: January 14, 2019. Accepted: July 19, 2019. Pre-published: August 1, 2019. Correspondence: STEFAN SCHEDING - [email protected] Li et al.: Supplemental data 1. Supplemental Materials and Methods BM-MNC isolation Bone marrow mononuclear cells (BM-MNC) from BM aspiration samples were isolated by density gradient centrifugation (LSM 1077 Lymphocyte, PAA, Pasching, Austria) either with or without prior incubation with RosetteSep Human Mesenchymal Stem Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) for lineage depletion (CD3, CD14, CD19, CD38, CD66b, glycophorin A). BM-MNCs from fetal long bones and adult hip bones were isolated as reported previously 1 by gently crushing bones (femora, tibiae, fibulae, humeri, radii and ulna) in PBS+0.5% FCS subsequent passing of the cell suspension through a 40-µm filter. -

Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Integration of 198 ChIP-seq datasets reveals human cis-regulatory regions Hamid Bolouri & Larry Ruzzo Thanks to Stephen Tapscott, Steven Henikoff & Zizhen Yao Slides will be available from: http://labs.fhcrc.org/bolouri/ Email [email protected] for manuscript (Bolouri & Ruzzo, submitted) Kleinjan & van Heyningen, Am. J. Hum. Genet., 2005, (76)8–32 Epstein, Briefings in Func. Genom. & Protemoics, 2009, 8(4)310-16 Regulation of SPi1 (Sfpi1, PU.1 protein) expression – part 1 miR155*, miR569# ~750nt promoter ~250nt promoter The antisense RNA • causes translational stalling • has its own promoter • requires distal SPI1 enhancer • is transcribed with/without SPI1. # Hikami et al, Arthritis & Rheumatism, 2011, 63(3):755–763 * Vigorito et al, 2007, Immunity 27, 847–859 Ebralidze et al, Genes & Development, 2008, 22: 2085-2092. Regulation of SPi1 expression – part 2 (mouse coordinates) Bidirectional ncRNA transcription proportional to PU.1 expression PU.1/ELF1/FLI1/GLI1 GATA1 GATA1 Sox4/TCF/LEF PU.1 RUNX1 SP1 RUNX1 RUNX1 SP1 ELF1 NF-kB SATB1 IKAROS PU.1 cJun/CEBP OCT1 cJun/CEBP 500b 500b 500b 500b 500b 750b 500b -18Kb -14Kb -12Kb -10Kb -9Kb Chou et al, Blood, 2009, 114: 983-994 Hoogenkamp et al, Molecular & Cellular Biology, 2007, 27(21):7425-7438 Zarnegar & Rothenberg, 2010, Mol. & cell Biol. 4922-4939 An NF-kB binding-site variant in the SPI1 URE reduces PU.1 expression & is GGGCCTCCCC correlated with AML GGGTCTTCCC Bonadies et al, Oncogene, 2009, 29(7):1062-72. SATB1 binding site A distal single nucleotide polymorphism alters long- range regulation of the PU.1 gene in acute myeloid leukemia Steidl et al, J Clin Invest. -

Molecular Codes for Cell Type Specification in Brn3 Retinal
Molecular codes for cell type specification in PNAS PLUS Brn3 retinal ganglion cells Szilard Sajgoa,1, Miruna Georgiana Ghiniaa,2, Matthew Brooksb, Friedrich Kretschmera,3, Katherine Chuanga,4, Suja Hiriyannac, Zhijian Wuc, Octavian Popescud,e, and Tudor Constantin Badeaa,5 aRetinal Circuits Development and Genetics Unit, Neurobiology–Neurodegeneration and Repair Laboratory, National Eye Institute, Bethesda, MD 20892; bGenomics Core, Neurobiology–Neurodegeneration and Repair Laboratory, National Eye Institute, Bethesda, MD 20892; cOcular Gene Therapy Core, National Eye Institute, Bethesda, MD 20892; dInstitute of Biology, Romanian Academy, Bucharest 060031, Romania; and eMolecular Biology Center, Interdisciplinary Research Institute on Bio-Nano-Science, Babes-Bolyai University, Cluj-Napoca 400084, Romania Edited by Jeremy Nathans, Johns Hopkins University, Baltimore, MD, and approved April 12, 2017 (received for review November 8, 2016) Visual information is conveyed from the eye to the brain by Onecut2 (6, 13, 26–33). Together with Isl1 and Brn3b, these distinct types of retinal ganglion cells (RGCs). It is largely unknown downstream factors are expressed in partially overlapping patterns how RGCs acquire their defining morphological and physiological in RGC types, and some were shown to be required for survival features and connect to upstream and downstream synaptic and/or dendrite and axon formation in various RGC types. partners. The three Brn3/Pou4f transcription factors (TFs) participate However, many other TFs may be involved -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Methodology article Open Access "NeuroStem Chip": a novel highly specialized tool to study neural differentiation pathways in human stem cells Sergey V Anisimov*, Nicolaj S Christophersen, Ana S Correia, Jia-Yi Li and Patrik Brundin Address: Neuronal Survival Unit, Wallenberg Neuroscience Center, Lund University, 221 84 Lund, Sweden Email: Sergey V Anisimov* - [email protected]; Nicolaj S Christophersen - [email protected]; Ana S Correia - [email protected]; Jia-Yi Li - [email protected]; Patrik Brundin - [email protected] * Corresponding author Published: 8 February 2007 Received: 21 September 2006 Accepted: 8 February 2007 BMC Genomics 2007, 8:46 doi:10.1186/1471-2164-8-46 This article is available from: http://www.biomedcentral.com/1471-2164/8/46 © 2007 Anisimov et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Human stem cells are viewed as a possible source of neurons for a cell-based therapy of neurodegenerative disorders, such as Parkinson's disease. Several protocols that generate different types of neurons from human stem cells (hSCs) have been developed. Nevertheless, the cellular mechanisms that underlie the development of neurons in vitro as they are subjected to the specific differentiation protocols are often poorly understood. Results: We have designed a focused DNA (oligonucleotide-based) large-scale microarray platform (named "NeuroStem Chip") and used it to study gene expression patterns in hSCs as they differentiate into neurons. -

Prostate News
Prostate Cancer and Prostatic Diseases (2005) 8, 2–6 & 2005 Nature Publishing Group All rights reserved 1365-7852/05 $30.00 www.nature.com/pcan Research Highlights Prostate News Prostate Cancer and Prostatic Diseases (2005) 8, 2–6. doi:10.1038/sj.pcan.4500793 Pokemon’s central role in cancer progression of prostate cancer. Epigenetic changes are reversible, which make them attractive targets for cancer development treatment with modulators that demethylate DNA and Researchers at New York’s Memorial Sloan-Kettering inhibit histone deacetylases, leading to reactivation of Cancer Center report novel findings in Nature regarding silenced genes. DNA methylation and histone modifica- a new cellular oncogene implicated in the development tions are important epigenetic mechanisms of gene of human cancers, including prostate neoplasia. Poke- regulation, and both play essential roles in tumor mon, a pop-culture inspired acronym for ‘POK erythroid initiation and progression. In prostate cancer, aberrant myeloid ontogenic factor’, is a POK protein family epigenetic events such as DNA hypo- and hypermethy- member encoded by the ZbTb7 gene and characterized lation and altered histone acetylation have been by its critical transcription function in cellular differ- observed to affect a large number of genes. However, entiation. Takahiro Maeda, Pier Paolo Pandolfi and no plenitude is without peril: although the number of colleagues show that Pokemon acts as a genuine proto- genes that undergo aberrant epigenetic inactivation oncogene when overexpressed by inhibiting the expres- associated with prostate cancer seems to be growing, sion of tumor suppressors, such as ARF. very few of these genes have yielded promising results To establish this novel finding, the authors conducted as potential tumor biomarkers for early diagnosis and a complete battery of genetic, clinical, and pathological risk assessment. -

FARE2021WINNERS Sorted by Institute
FARE2021WINNERS Sorted By Institute Swati Shah Postdoctoral Fellow CC Radiology/Imaging/PET and Neuroimaging Characterization of CNS involvement in Ebola-Infected Macaques using Magnetic Resonance Imaging, 18F-FDG PET and Immunohistology The Ebola (EBOV) virus outbreak in Western Africa resulted in residual neurologic abnormalities in survivors. Many case studies detected EBOV in the CSF, suggesting that the neurologic sequelae in survivors is related to viral presence. In the periphery, EBOV infects endothelial cells and triggers a “cytokine stormâ€. However, it is unclear whether a similar process occurs in the brain, with secondary neuroinflammation, neuronal loss and blood-brain barrier (BBB) compromise, eventually leading to lasting neurological damage. We have used in vivo imaging and post-necropsy immunostaining to elucidate the CNS pathophysiology in Rhesus macaques infected with EBOV (Makona). Whole brain MRI with T1 relaxometry (pre- and post-contrast) and FDG-PET were performed to monitor the progression of disease in two cohorts of EBOV infected macaques from baseline to terminal endpoint (day 5-6). Post-necropsy, multiplex fluorescence immunohistochemical (MF-IHC) staining for various cellular markers in the thalamus and brainstem was performed. Serial blood and CSF samples were collected to assess disease progression. The linear mixed effect model was used for statistical analysis. Post-infection, we first detected EBOV in the serum (day 3) and CSF (day 4) with dramatic increases until euthanasia. The standard uptake values of FDG-PET relative to whole brain uptake (SUVr) in the midbrain, pons, and thalamus increased significantly over time (p<0.01) and positively correlated with blood viremia (p≤0.01).