Estimating Gene Regulatory Activity Using Mathematical Optimization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Order Estimation Based Approach to Identify Response Genes
AN ORDER ESTIMATION BASED APPROACH TO IDENTIFY RESPONSE GENES FOR MICRO ARRAY TIME COURSE DATA A Thesis Presented to The Faculty of Graduate Studies of The University of Guelph by ZHIHENG LU In partial fulfilment of requirements for the degree of Doctor of Philosophy September, 2008 © Zhiheng Lu, 2008 Library and Bibliotheque et 1*1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de I'edition 395 Wellington Street 395, rue Wellington Ottawa ON K1A0N4 Ottawa ON K1A0N4 Canada Canada Your file Votre reference ISBN: 978-0-494-47605-5 Our file Notre reference ISBN: 978-0-494-47605-5 NOTICE: AVIS: The author has granted a non L'auteur a accorde une licence non exclusive exclusive license allowing Library permettant a la Bibliotheque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par telecommunication ou par Plntemet, prefer, telecommunication or on the Internet, distribuer et vendre des theses partout dans loan, distribute and sell theses le monde, a des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, electronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriete du droit d'auteur ownership and moral rights in et des droits moraux qui protege cette these. this thesis. Neither the thesis Ni la these ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent etre imprimes ou autrement may be printed or otherwise reproduits sans son autorisation. -
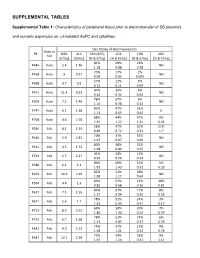
Supplemental Tables
SUPPLEMENTAL TABLES Supplemental Table 1: Characteristics of peripheral blood prior to electrotransfer of SB plasmids and numeric expansion on γ-irradiated AaPC and cytokines Day 0 (Day of electroporation) Auto or P# WBC ALC CD3 (ATC) CD4 CD8 ABC Allo [K/mL] [K/mL] [% & K/mL] [% & K/mL] [% & K/mL] [% & K/mL] 81% 60% 19% P446 Auto 5.4 1.46 ND 1.18 0.88 0.28 23% 17% 2% P458 Auto 9 0.27 ND 0.06 0.05 0.005 17% 12% 5% P468 Auto 4.7 0.9 ND 0.15 0.11 0.04 47% 35% 5% P471 Auto 11.4 0.93 ND 0.44 0.32 0.04 78% 67% 9% P509 Auto 7.2 1.46 ND 1.14 0.98 0.13 82% 47% 32% P747 Auto 6.1 1.38 0 1.13 0.65 0.44 88% 44% 47% 0% P708 Auto 4.6 2.78 2.45 1.22 1.31 0.05 58% 47% 20% 21% P396 Allo 8.1 1.54 0.89 0.72 0.31 1.7 70% 31% 32% P410 Allo 5.9 2.81 ND 1.97 0.87 0.90 80% 48% 32% P411 Allo 4.5 1.72 ND 1.38 0.83 0.55 41% 24% 15% P513 Allo 6.7 2.27 ND 0.93 0.54 0.34 86% 68% 15% 5% P580 Allo 6.1 2.1 1.81 1.43 0.32 0.29 82% 51% 28% P459 Allo 10.6 2.29 ND 1.88 1.17 0.64 64% 52% 12% 18% P564 Allo 4.4 1.3 0.83 0.68 0.16 0.81 82% 61% 17% 8% P617 Allo 7.1 1.55 1.27 0.94 0.26 0.58 78% 53% 24% 3% P671 Allo 5.4 1.7 1.33 0.90 0.41 0.17 69% 50% 20% 7% P723 Allo 8.6 2.61 1.80 1.30 0.52 0.59 78% 52% 22% 6% P732 Allo 6.7 1.68 1.31 0.87 0.37 0.39 74% 57% 15% 9% P641 Allo 9.0 2.13 1.58 1.21 0.32 0.78 73% 54% 18% 9% P647 Allo 12.1 2.29 1.67 1.24 0.41 1.11 75% 59% 11% P716 Allo 3.0 1.83 0 1.37 1.08 0.20 87% 73% 12% 1% P718 Allo 6.2 1.99 1.73 1.45 0.24 0.07 83% 69% 13% 6% P771 Allo 13.1 3.23 2.68 2.23 0.42 0.79 73% 42% 27% 6% P783 Allo 8.1 1.54 1.12 0.65 0.42 0.52 WBC = white blood -

Two Locus Inheritance of Non-Syndromic Midline Craniosynostosis Via Rare SMAD6 and 4 Common BMP2 Alleles 5 6 Andrew T
1 2 3 Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and 4 common BMP2 alleles 5 6 Andrew T. Timberlake1-3, Jungmin Choi1,2, Samir Zaidi1,2, Qiongshi Lu4, Carol Nelson- 7 Williams1,2, Eric D. Brooks3, Kaya Bilguvar1,5, Irina Tikhonova5, Shrikant Mane1,5, Jenny F. 8 Yang3, Rajendra Sawh-Martinez3, Sarah Persing3, Elizabeth G. Zellner3, Erin Loring1,2,5, Carolyn 9 Chuang3, Amy Galm6, Peter W. Hashim3, Derek M. Steinbacher3, Michael L. DiLuna7, Charles 10 C. Duncan7, Kevin A. Pelphrey8, Hongyu Zhao4, John A. Persing3, Richard P. Lifton1,2,5,9 11 12 1Department of Genetics, Yale University School of Medicine, New Haven, CT, USA 13 2Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT, USA 14 3Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT, USA 15 4Department of Biostatistics, Yale University School of Medicine, New Haven, CT, USA 16 5Yale Center for Genome Analysis, New Haven, CT, USA 17 6Craniosynostosis and Positional Plagiocephaly Support, New York, NY, USA 18 7Department of Neurosurgery, Yale University School of Medicine, New Haven, CT, USA 19 8Child Study Center, Yale University School of Medicine, New Haven, CT, USA 20 9The Rockefeller University, New York, NY, USA 21 22 ABSTRACT 23 Premature fusion of the cranial sutures (craniosynostosis), affecting 1 in 2,000 24 newborns, is treated surgically in infancy to prevent adverse neurologic outcomes. To 25 identify mutations contributing to common non-syndromic midline (sagittal and metopic) 26 craniosynostosis, we performed exome sequencing of 132 parent-offspring trios and 59 27 additional probands. -

P53 — 30 Yreviewsears On
FOCUS ON P53 — 30 YREVIEWSEARS ON p53 — a Jack of all trades but master of none Melissa R. Junttila and Gerard I. Evan Abstract | Cancers are rare because their evolution is actively restrained by a range of tumour suppressors. Of these p53 seems unusually crucial as either it or its attendant upstream or downstream pathways are inactivated in virtually all cancers. p53 is an evolutionarily ancient coordinator of metazoan stress responses. Its role in tumour suppression is likely to be a relatively recent adaptation, which is only necessary when large, long-lived organisms acquired the sufficient size and somatic regenerative capacity to necessitate specific mechanisms to reign in rogue proliferating cells. However, such evolutionary reappropriation of this venerable transcription factor entails compromises that restrict its efficacy as a tumour suppressor. Cnidarians–bilaterians Cancer is a genetic pathology that arises in the adult tissues questions are important for several reasons. Knowing Cinidarians comprise an animal of long-lived organisms, such as vertebrates, whose tis- how p53 discriminates between normal and tumour cells phylum of ~9,000 radially sues retain a substantial regenerative capacity throughout might point to attributes of cancer cells that qualitatively symmetrical, mostly marine life and, consequently, whose somatic cells accumulate distinguish them from normal somatic cells and could organisms. Most other animals are bilaterally symmetrical mutations. Occasionally, such mutations corrupt the be used as tumour-specific targets. Knowing what sig- and are classed as bilateria. regulatory mechanisms that suppress untoward somatic nals drive selection for loss of p53 function in cancers The cnidarians and bilaterians cell proliferation, survival and migration, resulting in the would help us understand what constrains and dictates last shared a common ancestor progressive outgrowth of a somatic clone. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Deregulated Mirnas in Bone Health Epigenetic Roles in Osteoporosis
Bone 122 (2019) 52–75 Contents lists available at ScienceDirect Bone journal homepage: www.elsevier.com/locate/bone Review Article Deregulated miRNAs in bone health: Epigenetic roles in osteoporosis T ⁎ D. Bellaviaa, , A. De Lucaa, V. Carinaa, V. Costaa, L. Raimondia, F. Salamannab, R. Alessandroc,d, M. Finib, G. Giavaresib a IRCCS Istituto Ortopedico Rizzoli, Bologna, Italy b IRCCS Istituto Ortopedico Rizzoli, Laboratory of Preclinical and Surgical Studies, Bologna, Italy c Department of Biopathology and Medical Biotechnologies, Section of Biology and Genetics, University of Palermo, Palermo 90133, Italy d Institute of Biomedicine and Molecular Immunology (IBIM), National Research Council, Palermo, Italy. ARTICLE INFO ABSTRACT Keywords: MicroRNA (miRNA) has shown to enhance or inhibit cell proliferation, differentiation and activity of different miRNA cell types in bone tissue. The discovery of miRNA actions and their targets has helped to identify them as novel Osteoporosis regulations actors in bone. Various studies have shown that miRNA deregulation mediates the progression of Osteogenesis bone-related pathologies, such as osteoporosis. Osteoblast The present review intends to give an exhaustive overview of miRNAs with experimentally validated targets Osteoclast involved in bone homeostasis and highlight their possible role in osteoporosis development. Moreover, the review analyzes miRNAs identified in clinical trials and involved in osteoporosis. 1. Introduction degradation [7]. Mature miRNAs are generated by the sequential cleavage of precursor transcripts, indicated as pri-miRNAs, that is Bone is a mineralized connective tissue that has two essential bio- cleaved in the nucleus by Drosha and the Di George syndrome critical logical roles: 1) a mechanical role – supporting body locomotion and region gene 8 (DGCR8) complex, producing a hairpin of 70–100 nu- protecting brain and splanchnic organs; and 2) a metabolic role - reg- cleotides, named pre-miRNA. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Greg's Awesome Thesis
Analysis of alignment error and sitewise constraint in mammalian comparative genomics Gregory Jordan European Bioinformatics Institute University of Cambridge A dissertation submitted for the degree of Doctor of Philosophy November 30, 2011 To my parents, who kept us thinking and playing This dissertation is the result of my own work and includes nothing which is the out- come of work done in collaboration except where specifically indicated in the text and acknowledgements. This dissertation is not substantially the same as any I have submitted for a degree, diploma or other qualification at any other university, and no part has already been, or is currently being submitted for any degree, diploma or other qualification. This dissertation does not exceed the specified length limit of 60,000 words as defined by the Biology Degree Committee. November 30, 2011 Gregory Jordan ii Analysis of alignment error and sitewise constraint in mammalian comparative genomics Summary Gregory Jordan November 30, 2011 Darwin College Insight into the evolution of protein-coding genes can be gained from the use of phylogenetic codon models. Recently sequenced mammalian genomes and powerful analysis methods developed over the past decade provide the potential to globally measure the impact of natural selection on pro- tein sequences at a fine scale. The detection of positive selection in particular is of great interest, with relevance to the study of host-parasite conflicts, immune system evolution and adaptive dif- ferences between species. This thesis examines the performance of methods for detecting positive selection first with a series of simulation experiments, and then with two empirical studies in mammals and primates. -

Functional Genomics Analysis of Vitamin D Effects on CD4+ T Cells In
Functional genomics analysis of vitamin D effects PNAS PLUS on CD4+ T cells in vivo in experimental autoimmune encephalomyelitis Manuel Zeitelhofera,b, Milena Z. Adzemovica, David Gomez-Cabreroc,d,e, Petra Bergmana, Sonja Hochmeisterf, Marie N’diayea, Atul Paulsona, Sabrina Ruhrmanna, Malin Almgrena, Jesper N. Tegnérc,d,g, Tomas J. Ekströma, André Ortlieb Guerreiro-Cacaisa, and Maja Jagodica,1 aDepartment of Clinical Neuroscience, Center for Molecular Medicine, Karolinska Institutet, 171 76 Stockholm, Sweden; bVascular Biology Unit, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, 171 77 Stockholm, Sweden; cUnit of Computational Medicine, Department of Medicine, Solna, Center for Molecular Medicine, Karolinska Institutet, 171 76 Stockholm, Sweden; dScience for Life Laboratory, 171 21 Solna, Sweden; eMucosal and Salivary Biology Division, King’s College London Dental Institute, London SE1 9RT, United Kingdom; fDepartment of General Neurology, Medical University of Graz, 8036 Graz, Austria; and gBiological and Environmental Sciences and Engineering Division, Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology, 23955 Thuwal, Kingdom of Saudi Arabia Edited by Tomas G. M. Hokfelt, Karolinska Institutet, Stockholm, Sweden, and approved January 19, 2017 (received for review September 24, 2016) Vitamin D exerts multiple immunomodulatory functions and has autoimmune destruction of myelin, axonal loss, and brain atro- been implicated in the etiology and treatment of several autoim- phy (6). Increased risk of developing MS has been described in mune diseases, including multiple sclerosis (MS). We have previously carriers of rare and common variants of the CYP27B gene (7, 8), reported that in juvenile/adolescent rats, vitamin D supplementation which encodes the enzyme that catalyzes the last step in con- protects from experimental autoimmune encephalomyelitis (EAE), a verting vitamin D to its active form, from 25(OH)D3 to 1,25 model of MS. -

Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Integration of 198 ChIP-seq datasets reveals human cis-regulatory regions Hamid Bolouri & Larry Ruzzo Thanks to Stephen Tapscott, Steven Henikoff & Zizhen Yao Slides will be available from: http://labs.fhcrc.org/bolouri/ Email [email protected] for manuscript (Bolouri & Ruzzo, submitted) Kleinjan & van Heyningen, Am. J. Hum. Genet., 2005, (76)8–32 Epstein, Briefings in Func. Genom. & Protemoics, 2009, 8(4)310-16 Regulation of SPi1 (Sfpi1, PU.1 protein) expression – part 1 miR155*, miR569# ~750nt promoter ~250nt promoter The antisense RNA • causes translational stalling • has its own promoter • requires distal SPI1 enhancer • is transcribed with/without SPI1. # Hikami et al, Arthritis & Rheumatism, 2011, 63(3):755–763 * Vigorito et al, 2007, Immunity 27, 847–859 Ebralidze et al, Genes & Development, 2008, 22: 2085-2092. Regulation of SPi1 expression – part 2 (mouse coordinates) Bidirectional ncRNA transcription proportional to PU.1 expression PU.1/ELF1/FLI1/GLI1 GATA1 GATA1 Sox4/TCF/LEF PU.1 RUNX1 SP1 RUNX1 RUNX1 SP1 ELF1 NF-kB SATB1 IKAROS PU.1 cJun/CEBP OCT1 cJun/CEBP 500b 500b 500b 500b 500b 750b 500b -18Kb -14Kb -12Kb -10Kb -9Kb Chou et al, Blood, 2009, 114: 983-994 Hoogenkamp et al, Molecular & Cellular Biology, 2007, 27(21):7425-7438 Zarnegar & Rothenberg, 2010, Mol. & cell Biol. 4922-4939 An NF-kB binding-site variant in the SPI1 URE reduces PU.1 expression & is GGGCCTCCCC correlated with AML GGGTCTTCCC Bonadies et al, Oncogene, 2009, 29(7):1062-72. SATB1 binding site A distal single nucleotide polymorphism alters long- range regulation of the PU.1 gene in acute myeloid leukemia Steidl et al, J Clin Invest. -

BMC Genomics Biomed Central
BMC Genomics BioMed Central Methodology article Open Access "NeuroStem Chip": a novel highly specialized tool to study neural differentiation pathways in human stem cells Sergey V Anisimov*, Nicolaj S Christophersen, Ana S Correia, Jia-Yi Li and Patrik Brundin Address: Neuronal Survival Unit, Wallenberg Neuroscience Center, Lund University, 221 84 Lund, Sweden Email: Sergey V Anisimov* - [email protected]; Nicolaj S Christophersen - [email protected]; Ana S Correia - [email protected]; Jia-Yi Li - [email protected]; Patrik Brundin - [email protected] * Corresponding author Published: 8 February 2007 Received: 21 September 2006 Accepted: 8 February 2007 BMC Genomics 2007, 8:46 doi:10.1186/1471-2164-8-46 This article is available from: http://www.biomedcentral.com/1471-2164/8/46 © 2007 Anisimov et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Human stem cells are viewed as a possible source of neurons for a cell-based therapy of neurodegenerative disorders, such as Parkinson's disease. Several protocols that generate different types of neurons from human stem cells (hSCs) have been developed. Nevertheless, the cellular mechanisms that underlie the development of neurons in vitro as they are subjected to the specific differentiation protocols are often poorly understood. Results: We have designed a focused DNA (oligonucleotide-based) large-scale microarray platform (named "NeuroStem Chip") and used it to study gene expression patterns in hSCs as they differentiate into neurons. -

Involvement of Human Micro-RNA in Growth and Response to Chemotherapy in Human Cholangiocarcinoma Cell Lines
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Texas A&M Repository GASTROENTEROLOGY 2006;130:2113–2129 Involvement of Human Micro-RNA in Growth and Response to Chemotherapy in Human Cholangiocarcinoma Cell Lines FANYIN MENG,* ROGER HENSON,* MOLLY LANG,* HANIA WEHBE,* SHAIL MAHESHWARI,* JOSHUA T. MENDELL,‡ JINMAI JIANG,§ THOMAS D. SCHMITTGEN,§ and TUSHAR PATEL* *Department of Internal Medicine, Scott and White Clinic, Texas A&M University System Health Science Center College of Medicine, Temple, Texas; ‡The Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland; and §College of Pharmacy, The Ohio State University, Columbus, Ohio Background & Aims: Micro-RNA (miRNA) are endoge- age of target mRNA. Although several hundred miRNA nous regulatory RNA molecules that modulate gene have been cloned or predicted, a functional role for these expression. Alterations in miRNA expression can contrib- in many cellular processes has not been fully elucidated.1 ute to tumor growth by modulating the functional ex- The best characterized role of miRNA has been as a pression of critical genes involved in tumor cell prolifer- developmental regulator in Caenorhabditis elegans and ation or survival. Our aims were to identify specific Drosophilia. More recently, several other roles have been miRNA involved in the regulation of cholangiocarcinoma described including regulation of apoptosis and differen- growth and response to chemotherapy. Methods: miRNA tiation. miRNA are expressed as long precursor RNA expression in malignant and nonmalignant human that are processed by a single nuclease Drosha prior to cholangiocytes was assessed using a microarray. Ex- pression of selected miRNA and their precursors was transport into the cytoplasm by an exportin-5-dependent evaluated by Northern blots and real-time polymerase mechanism.