Prevalence of Chromosomal Rearrangements Involving Non-ETS Genes in Prostate Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Divergence of the Genes on Human Chromosome 21 Between Human and Other Hominoids and Variation of Substitution Rates Among Transcription Units
Divergence of the genes on human chromosome 21 between human and other hominoids and variation of substitution rates among transcription units Jinxiu Shi*†‡, Huifeng Xi*‡§, Ying Wang*, Chenghui Zhang*, Zhengwen Jiang§¶, Kuixing Zhang*, Yayun Shen*, Lin Jin*, Kaiyue Zhang*, Wentao Yuan*, Ying Wang*, Jie Lin*, Qi Hua*, Fengqing Wang*, Shuhua Xu*, Suangxi Ren*, Shijie Xu*†, Guoping Zhao*, Zhu Chen*†§, Li Jin*§¶ʈ, and Wei Huang*†ʈ *Chinese National Human Genome Center at Shanghai, 250 Bi Bo Road, Shanghai 201203, People’s Republic of China; †Health Science Center, Shanghai Second Medical University and Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 225 Chongqing Nan Road, Shanghai 200025, People’s Republic of China; §Morgan-Tan International Center for Life Science and Center for Anthropological Sciences, School of Biological Sciences, Fudan University, 220 Han Dan Road, Shanghai 200433, People’s Republic of China; and ¶Center for Genome Information, Department of Environmental Health, University of Cincinnati College of Medicine, P.O. Box 670056, Cincinnati, OH 45267-0056 Communicated by Jiazhen Tan, Fudan University, Shanghai, People’s Republic of China, May 5, 2003 (received for review December 16, 2002) The study of genomic divergence between humans and primates of human beings and a pool of 20 chimpanzee samples, which may provide insight into the origins of human beings and the allows a direct comparison of the genomes of these two species. genetic basis of unique human traits and diseases. Chromosome 21 As outgroups, partial sequences of those genes were also deter- is the smallest chromosome in the human genome, and some of its mined for a gorilla, an orangutan, and a macaque. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Exploring Prostate Cancer Genome Reveals Simultaneous Losses of PTEN, FAS and PAPSS2 in Patients with PSA Recurrence After Radical Prostatectomy
Int. J. Mol. Sci. 2015, 16, 3856-3869; doi:10.3390/ijms16023856 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article Exploring Prostate Cancer Genome Reveals Simultaneous Losses of PTEN, FAS and PAPSS2 in Patients with PSA Recurrence after Radical Prostatectomy Chinyere Ibeawuchi 1, Hartmut Schmidt 2, Reinhard Voss 3, Ulf Titze 4, Mahmoud Abbas 5, Joerg Neumann 6, Elke Eltze 7, Agnes Marije Hoogland 8, Guido Jenster 9, Burkhard Brandt 10 and Axel Semjonow 1,* 1 Prostate Center, Department of Urology, University Hospital Muenster, Albert-Schweitzer-Campus 1, Gebaeude 1A, Muenster D-48149, Germany; E-Mail: [email protected] 2 Center for Laboratory Medicine, University Hospital Muenster, Albert-Schweitzer-Campus 1, Gebaeude 1A, Muenster D-48149, Germany; E-Mail: [email protected] 3 Interdisciplinary Center for Clinical Research, University of Muenster, Albert-Schweitzer-Campus 1, Gebaeude D3, Domagkstrasse 3, Muenster D-48149, Germany; E-Mail: [email protected] 4 Pathology, Lippe Hospital Detmold, Röntgenstrasse 18, Detmold D-32756, Germany; E-Mail: [email protected] 5 Institute of Pathology, Mathias-Spital-Rheine, Frankenburg Street 31, Rheine D-48431, Germany; E-Mail: [email protected] 6 Institute of Pathology, Klinikum Osnabrueck, Am Finkenhuegel 1, Osnabrueck D-49076, Germany; E-Mail: [email protected] 7 Institute of Pathology, Saarbrücken-Rastpfuhl, Rheinstrasse 2, Saarbrücken D-66113, Germany; E-Mail: [email protected] 8 Department -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -

ARTICLE Doi:10.1038/Nature10523
ARTICLE doi:10.1038/nature10523 Spatio-temporal transcriptome of the human brain Hyo Jung Kang1*, Yuka Imamura Kawasawa1*, Feng Cheng1*, Ying Zhu1*, Xuming Xu1*, Mingfeng Li1*, Andre´ M. M. Sousa1,2, Mihovil Pletikos1,3, Kyle A. Meyer1, Goran Sedmak1,3, Tobias Guennel4, Yurae Shin1, Matthew B. Johnson1,Zˇeljka Krsnik1, Simone Mayer1,5, Sofia Fertuzinhos1, Sheila Umlauf6, Steven N. Lisgo7, Alexander Vortmeyer8, Daniel R. Weinberger9, Shrikant Mane6, Thomas M. Hyde9,10, Anita Huttner8, Mark Reimers4, Joel E. Kleinman9 & Nenad Sˇestan1 Brain development and function depend on the precise regulation of gene expression. However, our understanding of the complexity and dynamics of the transcriptome of the human brain is incomplete. Here we report the generation and analysis of exon-level transcriptome and associated genotyping data, representing males and females of different ethnicities, from multiple brain regions and neocortical areas of developing and adult post-mortem human brains. We found that 86 per cent of the genes analysed were expressed, and that 90 per cent of these were differentially regulated at the whole-transcript or exon level across brain regions and/or time. The majority of these spatio-temporal differences were detected before birth, with subsequent increases in the similarity among regional transcriptomes. The transcriptome is organized into distinct co-expression networks, and shows sex-biased gene expression and exon usage. We also profiled trajectories of genes associated with neurobiological categories and diseases, and identified associations between single nucleotide polymorphisms and gene expression. This study provides a comprehensive data set on the human brain transcriptome and insights into the transcriptional foundations of human neurodevelopment. -

Bayesian GWAS and Network Analysis Revealed New Candidate Genes for Number of Teats in Pigs
J Appl Genetics DOI 10.1007/s13353-014-0240-y ANIMAL GENETICS • ORIGINAL PAPER Bayesian GWAS and network analysis revealed new candidate genes for number of teats in pigs L. L. Verardo & F. F. Silva & L. Varona & M. D. V. Resende & J. W. M. Bastiaansen & P. S. Lopes & S. E. F. Guimarães Received: 7 March 2014 /Revised: 27 May 2014 /Accepted: 23 July 2014 # Institute of Plant Genetics, Polish Academy of Sciences, Poznan 2014 Abstract The genetic improvement of reproductive traits comparisons based on deviance posterior distribution indicat- such as the number of teats is essential to the success of the ed the superiority of Gaussian model. In general, our results pig industry. As opposite to most SNP association studies that suggest the presence of 19 significant SNPs, which mapped consider continuous phenotypes under Gaussian assumptions, 13 genes. Besides, we predicted gene interactions through this trait is characterized as a discrete variable, which could networks that are consistent with the mammals known breast potentially follow other distributions, such as the Poisson. biology (e.g., development of prolactin receptor signaling, and Therefore, in order to access the complexity of a counting cell proliferation), captured known regulation binding sites, random regression considering all SNPs simultaneously as and provided candidate genes for that trait (e.g., TINAGL1 covariate under a GWAS modeling, the Bayesian inference and ICK). tools become necessary. Currently, another point that deserves to be highlighted in GWAS is the genetic dissection of com- Keywords Counting data . Genes . Reproductive traits . SNP plex phenotypes through candidate genes network derived association from significant SNPs. -
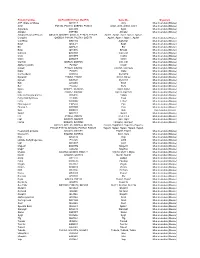
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Transcriptomic Signature and Metabolic Programming of Bovine Classical and Nonclassical Monocytes Indicate Distinct Functional Specializations
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.30.362731; this version posted November 1, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Transcriptomic signature and metabolic programming of bovine classical and nonclassical monocytes indicate distinct functional specializations Stephanie C. Talker1,2, G. Tuba Barut1,2, Reto Rufener3, Lilly von Münchow4, Artur Summerfield1,2 1Institute of Virology and Immunology, Bern and Mittelhäusern, Switzerland 2Department of Infectious Diseases and Pathobiology, Vetsuisse Faculty, University of Bern, Bern, Switzerland 3Institute of Parasitology, Vetsuisse Faculty, University of Bern, Bern, Switzerland 4 Bucher Biotec AG, Basel, Switzerland *Correspondence: Corresponding Author [email protected] Keywords: monocyte subsets, transcriptome, metabolism, cattle Abstract Similar to human monocytes, bovine monocytes can be split into CD14+CD16- classical and CD14-CD16+ nonclassical monocytes (cM and ncM, respectively). Here, we present an in-depth analysis of their steady-state transcriptomes, highlighting pronounced functional specializations. Gene transcription indicates that pro-inflammatory and antibacterial processes are associated with cM, while ncM appear to be specialized in regulatory/anti-inflammatory functions and tissue repair, as well as antiviral responses and T-cell immunomodulation. In support of these functional differences, we found that oxidative phosphorylation prevails in ncM, whereas cM are clearly biased towards aerobic glycolysis. Furthermore, bovine monocyte subsets differed in their responsiveness to TLR ligands, supporting an antiviral role of ncM. Taken together, these data clearly indicate a variety of subset-specific functions in cM and ncM that are likely to be transferable to monocyte subsets of other species, including humans. -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -

Dissecting the Non-Canonical Functions of P53 Through Novel Target Identification and P53 Acetylation
Dissecting the non-canonical functions of p53 through novel target identification and p53 acetylation Shang-Jui Wang Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Shang-Jui Wang All rights reserved ABSTRACT Dissecting the non-canonical functions of p53 through novel target identification and p53 acetylation Shang-Jui Wang It is well established that the p53 tumor suppressor plays a crucial role in controlling cell proliferation and apoptosis upon various types of stress. There is increasing evidence showing that p53 is also critically involved in various non-canonical pathways, including metabolism, autophagy, senescence and aging. Through a ChIP-on-chip screen, we identified a novel p53 metabolic target, pantothenate kinase-1 (PANK1). PanK1 catalyzes the rate-limiting step for CoA synthesis, and therefore, controls intracellular CoA content; Pank1 knockout mice exhibit defect in -oxidation and gluconeogenesis in the liver after starvation due to insufficient CoA levels. We demonstrated that PANK1 gene is a direct transcriptional target of p53. Although DNA damage-induced p53 upregulates PanK1 expression, depletion of PanK1 expression does not affect p53-dependent growth arrest or apoptosis. Interestingly, upon glucose starvation, PanK1 expression is significantly reduced in HCT116 p53 (-/-) but not in HCT116 p53 (+/+) cells, suggesting that p53 is required to maintain PanK1 expression under metabolic stress conditions. Moreover, by using p53-mutant mice, we observed that PanK activity and CoA levels are lower in livers of p53-null mice than that of wild-type mice upon starvation.