Interaction of REPS2 with NF-Κb in Prostate Cancer Cells
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The BCR/ABL Oncogene Alters Interaction of the Adapter Proteins CRKL and CRK with Cellular Proteins N Uemura, R Salgia, J-L Li, E Pisick, M Sattler and JD Griffin
Leukemia (1997) 11, 376–385 1997 Stockton Press All rights reserved 0887-6924/97 $12.00 The BCR/ABL oncogene alters interaction of the adapter proteins CRKL and CRK with cellular proteins N Uemura, R Salgia, J-L Li, E Pisick, M Sattler and JD Griffin Division of Hematologic Malignancies, Dana-Farber Cancer Institute and Harvard Medical School, 44 Binney Street, Boston, MA 02115, USA The Philadelphia chromosome translocation generates a chim- alternative splicing of the human CRK proto-oncogene.8,9 eric oncogene, BCR/ABL, which causes chronic myelogenous CRKL has the same overall organization as CRK II, consisting leukemia (CML). In primary leukemic neutrophils from patients of one N-terminal SH2 domain followed by two SH3 domains, with CML, the major tyrosine phosphorylated protein is CRKL, 9 an SH2-SH3-SH3 adapter protein which has an overall hom- and does not contain other known functional motifs. CRK I ology of 60% to CRK, the human homologue of the v-crk onco- lacks the C-terminal SH3 domain of CRK II and CRKL, and gene. In cell lines transformed by BCR/ABL, CRKL was tyrosine overexpression of CRK I, but not CRK II, leads to transform- phosphorylated, while CRK was not. We looked for changes in ation of mammalian fibroblasts.8 v-crk is the oncogene in the CRK- and CRKL-binding proteins in Ba/F3 hematopoietic cell avian retrovirus CT10, and relative to CRK, v-Crk has a lines which were transformed by BCR/ABL. Anti-CRK II or anti- deletion of the C-terminal SH3 domain and the major tyrosine CRKL immunoprecipitates were probed by far Western blotting 221 10 with CRK II- or CRKL-GST fusion proteins to display CRK- and phosphorylation site at tyr . -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -

Liquid-Like Protein Interactions Catalyze Assembly of Endocytic Vesicles
bioRxiv preprint doi: https://doi.org/10.1101/860684; this version posted December 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. **This manuscript is being posted in tandem with a manuscript by M. Kozak and M. Kaksonen which reports complementary findings in budding yeast. Title Liquid-like protein interactions catalyze assembly of endocytic vesicles Authors Kasey J. Daya, Grace Kagob, Liping Wangc, J Blair Richterc, Carl C. Haydena, Eileen M. Laferc, Jeanne C. Stachowiaka,b,* Affiliations a Department of Biomedical Engineering, The University of Texas at Austin, Austin, TX, 78712, USA; b Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, TX, 78712, USA; c Department of Biochemistry and Structural Biology, Center for Biomedical Neuroscience, The University of Texas Health Science Center at San Antonio, San Antonio, TX, 78229, USA * To whom correspondence should be addressed: [email protected]. 1 bioRxiv preprint doi: https://doi.org/10.1101/860684; this version posted December 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract During clathrin-mediated endocytosis, dozens of proteins assemble into an interconnected network at the plasma membrane. As early initiators of endocytosis, Eps15 and Fcho1 are responsible for locally concentrating downstream components on the membrane surface. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

RNF11 at the Crossroads of Protein Ubiquitination
biomolecules Review RNF11 at the Crossroads of Protein Ubiquitination Anna Mattioni, Luisa Castagnoli and Elena Santonico * Department of Biology, University of Rome Tor Vergata, Via della ricerca scientifica, 00133 Rome, Italy; [email protected] (A.M.); [email protected] (L.C.) * Correspondence: [email protected] Received: 29 September 2020; Accepted: 8 November 2020; Published: 11 November 2020 Abstract: RNF11 (Ring Finger Protein 11) is a 154 amino-acid long protein that contains a RING-H2 domain, whose sequence has remained substantially unchanged throughout vertebrate evolution. RNF11 has drawn attention as a modulator of protein degradation by HECT E3 ligases. Indeed, the large number of substrates that are regulated by HECT ligases, such as ITCH, SMURF1/2, WWP1/2, and NEDD4, and their role in turning off the signaling by ubiquitin-mediated degradation, candidates RNF11 as the master regulator of a plethora of signaling pathways. Starting from the analysis of the primary sequence motifs and from the list of RNF11 protein partners, we summarize the evidence implicating RNF11 as an important player in modulating ubiquitin-regulated processes that are involved in transforming growth factor beta (TGF-β), nuclear factor-κB (NF-κB), and Epidermal Growth Factor (EGF) signaling pathways. This connection appears to be particularly significant, since RNF11 is overexpressed in several tumors, even though its role as tumor growth inhibitor or promoter is still controversial. The review highlights the different facets and peculiarities of this unconventional small RING-E3 ligase and its implication in tumorigenesis, invasion, neuroinflammation, and cancer metastasis. Keywords: Ring Finger Protein 11; HECT ligases; ubiquitination 1. -

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -
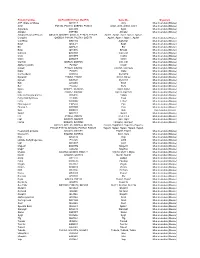
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse) -

Robust Identification of Somatic Mutations in Acute Myeloid Leukemia Using RNA-Seq
RNAmut: Robust Identification of Somatic Mutations in Acute Myeloid Leukemia Using RNA-seq Muxin Gu1,2, Maximillian Zwiebel1,3, Swee Hoe Ong4, Nick Boughton5, Josep Nomdedeu1,2,6, Faisal Basheer1,2,7, Yasuhito Nannya8, Pedro M. Quiros1,2, Seishi Ogawa8, Mario Cazzola9, Roland Rad3, Adam P. Butler4, MS Vijayabaskar1,2,* and George Vassiliou1,2,7,* 1. Haematological Cancer Genetics, Wellcome Sanger Institute, Hinxton, Cambridge, UK 2. Wellcome Trust–MRC Stem Cell Institute, Cambridge Biomedical Campus, University of Cambridge, Cambridge, UK 3. German Consortium for Translational Cancer Research (DKTK), Partnering Site, Munich, 81377 Munich, Germany 4. Cancer Ageing and Somatic Mutation, Wellcome Sanger Institute, Hinxton, Cambridge, UK 5. Core Software Services, Wellcome Sanger Institute, Hinxton, Cambridge, UK 6. Hospital de la Santa Creu I Sant Pau, Barcelona, Spain 7. Department of Haematology, Cambridge University Hospitals NHS Trust, Cambridge, UK 8. Department of Pathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Kyoto 606-8501, Japan 9. Fondazione IRCCS Policlinico San Matteo and University of Pavia, 27100 Pavia, Italy *Correspondence to: [email protected] and [email protected] Acute myeloid leukemia (AML) is an aggressive malignancy of haematopoietic stem cells driven by a well-defined set of somatic mutations1, 2. Identifying the mutations driving individual cases is important for assigning the patient to a recognized WHO category, establishing prognostic risk and tailoring post-consolidation therapy3. As a result, AML research and diagnostic laboratories apply diverse methodologies to detect important mutations and many are introducing next generation sequencing approaches to study extended panels of genes in order to refine genomic classification and prognostic category1. -

Datasheet: VPA00306 Product Details
Datasheet: VPA00306 Description: RABBIT ANTI EPN1 Specificity: EPN1 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 100 µl Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Product Form Purified IgG - liquid Preparation Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 203-232 of human EPN1 External Database Links UniProt: Q9Y6I3 Related reagents Entrez Gene: 29924 EPN1 Related reagents Specificity Rabbit anti Human EPN1 antibody recognizes EPN1, also known as EH domain-binding mitotic phosphoprotein, Epsin 1 or EPS-15-interacting protein 1. The protein encoded by the EPN1 gene binds clathrin and is involved in the endocytosis of clathrin- coated vesicles. Three transcript variants encoding different isoforms have been found for EPN1 Page 1 of 2 (provided by RefSeq, Nov 2011). Rabbit anti Human EPN1 antibody detects a band of 90 kDa. -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures.