Theoretical Study for High-Energy-Density Compounds Derived from Cyclophosphazene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

12. FORMULATION of the URBAN TRANSPORT MASTER PLAN Development of the RSTP Urban Transportation Master Plan (1) Methodology
The Project on The Revision and Updating of the Strategic Transport Plan for Dhaka (RSTP) Final Report 12. FORMULATION OF THE URBAN TRANSPORT MASTER PLAN Development of the RSTP Urban Transportation Master Plan (1) Methodology The development of the RSTP Urban Transportation Master Plan adopted the following methodology (see Figure 12.1): (i) Elaborate the master plan network through a screen line analysis by comparing the network capacity and future demand. (ii) Identify necessary projects to meet future demand at the same time avoiding excessive capacity. (iii) Conducts economic evaluation of each project to give priority on projects with higher economic return. (iv) Conduct preliminary environmental assessment of every project and consider countermeasures against environmental problems, if any. (v) Make a final prioritization of all physical projects by examining their respective characteristics from different perspectives. (vi) Classify the projects into three categories, namely short-, medium- and long-term projects, by considering the financial constraints. (vii) Prepare an action plan for short-term projects together with “soft” measures. Mid-term Project Source: RSTP Study Team Figure 12.1 Development Procedure for the Master Plan 12-1 The Project on The Revision and Updating of the Strategic Transport Plan for Dhaka (RSTP) Final Report (2) Output of the Transportation Network Plan The RSTP urban transportation network plan was developed based on a review and a modification of the STP network plan. The main points of the modification or adoption of the STP network master plan are as follows: i. Harmonization with future urban structure, land-use plan and development of network plan. -

Effect of Nitrogen and Boron on the Yield of Wheat Cv. Kanchan
J. Bangladesh Agril. Univ. 3(2): 215-220, 2005 ISSN 1810-3030 Effect of nitrogen and boron on the yield of wheat cv. Kanchan M.M. Khan', A.K. Hasan2, M.H. Rashid2, and F. Ahrned3 Palli Karma Sahayak Foundation (PKSF), Sher-e-Banglanagar, Dhaka, 2Department of Agronomy and 3 Department of Agroforestry, Bangladesh Agricultural University, Mymensingh Abstract An experiment was carried out at the Agronomy Field Laboratory, Bangladesh Agricultural University, Mymensingh during the period from January to April 2004 to study the effect of different levels of nitrogen and boron on the yield of wheat cv. Kanchan. The treatments included four levels of nitrogen VIZ., 45, 60, 85 and 110 kg N ha-1 and four levels of boron viz., 0, 1, 2 and 3 kg B ha-1. The experiment was laid out in a Randomized Complete Block Design with three replications. The results revealed that Yield and yield contributing characters were influenced significantly by both levels of nitrogen and boron. Among the levels of nitrogen, 110 kg N ha-1 produced the highest grain (5.54 t ha-1)and straw (8.21 t ha-1 ) yields. The lowest grain (3.23 t ha-1) and straw (5.52 t ha-1) yields were observed with the application of 45 kg N ha-1. The highest grain (4.95 t ha-1)and straw (7.38 t ha 1) yields were produced With 3 kg B ha-1. The minimum grain (3.59 t ha-1)and straw (6.14 t ha-1)yields were found in the control treatment. -

Connecting Bangladesh: Economic Corridor Network
Connecting Bangladesh: Economic Corridor Network Economic corridors are anchored on transport corridors, and international experience suggests that the higher the level of connectivity within and across countries, the higher the level of economic growth. In this paper, a new set of corridors is being proposed for Bangladesh—a nine-corridor comprehensive integrated multimodal economic corridor network resembling the London Tube map. This paper presents the initial results of the research undertaken as an early step of that development effort. It recommends an integrated approach to developing economic corridors in Bangladesh that would provide a strong economic foundation for the construction of world-class infrastructure that, in turn, could support the growth of local enterprises and attract foreign investment. About the Asian Development Bank COnnecTING BANGLADESH: ADB’s vision is an Asia and Pacific region free of poverty. Its mission is to help its developing member countries reduce poverty and improve the quality of life of their people. Despite the region’s many successes, it remains home to a large share of the world’s poor. ADB is committed to reducing poverty through inclusive economic growth, environmentally sustainable growth, and regional integration. ECONOMIC CORRIDOR Based in Manila, ADB is owned by 67 members, including 48 from the region. Its main instruments for helping its developing member countries are policy dialogue, loans, equity investments, guarantees, grants, NETWORK and technical assistance. Mohuiddin Alamgir -
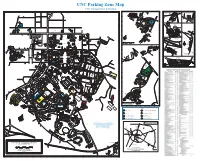
UNC Parking Zone Map UNC Transportation & Parking
UNC Parking Zone Map UNC Transportation & Parking Q R S T U V W X Y Z A B C D E F G H I J K L M N O P 26 **UNC LEASES SPACE CAROLINA . ROAD IN THESE BUILDINGS 21 21 MT HOMESTEAD NORTH LAND MGMT. PINEY OPERATIONS CTR. VD. (NC OFFICE HORACE WILLIAMS AIRPORT VD., HILL , JR. BL “RR” 41 1 1 Resident 41 CommuterRR Lot R12 UNC VD AND CHAPEL (XEROX) TE 40 MLK BL A PRINTING RIVE EXTENSION MLK BL ESTES D SERVICES TIN LUTHER KING TERST PLANT N O I AHEC T EHS HOMESTEAD ROAD MAR HANGER VD. 86) O I-40 STORAGE T R11 TH (SEE OTHER MAPS) 22 22 O 720, 725, & 730 MLK, JR. BL R1 T PHYSICAL NOR NORTH STREET ENVRNMEN HL .3 MILES TO TH. & SAFETY ESTES DRIVE 42 COMMUTER LOT T. 42 ER NC86 ELECTRICAL DISTRICENTBUTION OPERATIONS SURPLUS WA REHOUSE N1 ST GENERAL OREROOM 2 23 23 2 R1 CHAPEL HILL ES MLK JR. BOULE NORTH R1 ARKING ARD ILITI R1 / R2OVERFLOW ZONEP V VICES C R A F SHOPS GY SE EY 43 RN 43 ENERBUILDING CONSTRUCTION PRITCHARD STREET R1 NC 86 CHURCH STREET . HO , JR. BOULE ES F R1 / V STREET SER L BUILDING VICE ARD A ST ATIO GI EET N TR AIRPOR R2 S T DRIVE IN LUTHER KING BRANCH T L MAR HIL TH WEST ROSEMARY STREET EAST ROSEMARY STREET L R ACILITIES DRIVE F A NO 24 STUDRT 24 TH COLUMBI IO CHAPE R ADMINIST OFF R NO BUILDINGICE ATIVE R10 1700 N9 MLK 208 WEST 3 N10 FRANKLIN ST. -
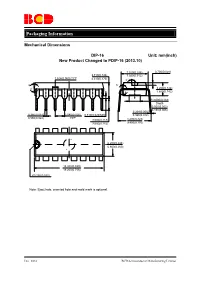
Mechanical-Dimensions-BCD.Pdf
Packaging Information Mechanical Dimensions DIP-16 Unit: mm(inch) New Product Changed to PDIP-16 (2013.10) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060) TYP 4.310(0.170) 5° 6° 6° 4° 3.200(0.126) 4° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014) 0.560(0.022) TYP 3.000(0.118) 8.200(0.323) 3.600(0.142) 9.400(0.370) 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optional. Dec. 2014 BCD Semiconductor Manufacturing Limited Packaging Information Mechanical Dimensions DIP-16 (Special for AZ4052) Unit: mm(inch) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060)T YP 4.310(0.170) 5 ° 6 ° 6 ° 4 ° 3.200(0.126) 4 ° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014 ) 0.560(0.022) TYP 3.000(0.118) 8.500(0.335) 3.600(0.142) TYP 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optiona.l Dec. -

Land Resource Appraisal of Bangladesh for Agricultural
BGD/81/035 Technical Report 3 Volume II LAND RESOURCES APPRAISAL OF BANGLADESH FOR AGRICULTURAL DEVELOPMENT REPORT 3 LAND RESOURCES DATA BASE VOLUME II SOIL, LANDFORM AND HYDROLOGICAL DATA BASE A /UNITED NATIONS DEVELOPMENT PROGRAMME FAo FOOD AND AGRICULTURE ORGANIZATION vJ OF THE UNITED NATIONS BGD/81/035 Technical Report 3 Volume II LAND RESOURCES APPRAISAL OF BANGLADESH FOR AGRICULTURALDEVELOPMENT REPORT 3 LAND RESOURCES DATA BASE VOLUME II SOIL, LANDFORM AND HYDROLOGICAL DATA BASE Report prepared for the Government of the People's Republic of Bangladesh by the Food and Agriculture Organization of the United Nations acting as executing agency for the United Nations Development Programme based on the work of H. Brammer Agricultural Development Adviser J. Antoine Data Base Management Expert and A.H. Kassam and H.T. van Velthuizen Land Resources and Agricultural Consultants UNITED NATIONS DEVELOPMENT PROGRAMME FOOD AND AGRICULTURE ORGANIZATION OF THE UNITED NATIONS Rome, 1988 The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Food and AgricultureOrganization of the United Nations concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. All rights reserved. No part of this publication may be reproduced, stored ina retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopyingor otherwise, without the prior perrnission of (he copyright owner. Applications for such permission,with a statement of the purpose and extent of the reproduction, should be addressedto the Director, Publications Division, Food and Agriculture Organization of the United Nations, Viadelle Terme di Caracarla, 00100 Home, Italy. -

Bangladesh Agron
Bangladesh Agron. J. 2015, 18(1): 89-98 GROWTH, YIELD AND QUALITY OF WHEAT VARITIES AS AFFECTED BY DIFFERENT LEVELS OF NITROGEN H. Mondal1, S. Mazumder2, S. K. Roy3, T. A. Mujahidi4 and S. K. Paul5 1Department of Agronomy, Sher-e-Bangla Agricultural University, Dhaka-1207, Bangladesh 2Sher-e-Bangla Nagar Adorsha Mohila College, Dhaka-1207, Bangladesh, 3Department of Genetics and Plant Breeding, Sher-e-Bangla Agricultural University, Dhaka-1207, Bangladesh 4Plant Breeding Division, Bangladesh Agricultural Research Institute, Joydebpur, Gazipur-1701, Bangladesh 5Agronomy Division, Bangladesh Agricultural Research Institute, Joydebpur, Gazipur-1701, Bangladesh. E-mail: [email protected] Key Words: Wheat variety, Nitrogen, Protein, Grain quality Abstract A field experiment was conducted at the experimental field of Sher-e-Bangla Agricultural University, Dhaka, Bangladesh during November 2012 to March 2013 to evaluate the response of three (3) wheat varieties viz., BARI Gom23, BARI Gom24 and BARI Gom25 under four levels of nitrogen fertilizer i.e, 75, 100, 125 and 150 kg N ha-1. The experiment was laid out in Randomized Complete Block Design (RCBD) with three replications. Results showed that plant height, number of leaves plant-1, leaf length and dry matter content were significantly affected due to varieties and/or nitrogen levels. Grains ear-1, number of fertile grains plant-1, 1000-grain weight, grain yield and harvest index were also significantly influenced by varieties and/or nitrogen levels. The value of all parameters studied in this experiment increased with increasing nitrogen levels up to 125 kg N ha-1 and thereafter decreased with fertilizer increasing level. Combination results showed that BARI Gom-24 with application of 125 kg N ha-1 gave the maximum grain yield (4.71 t ha-1), harvest index (49.37 %) and protein content (10.88%). -

ROAD SAFETY REPORTS Dhaka Metropolitan Police
ROAD SAFETY REPORTS Dhaka Metropolitan Police Traffic Accident Analysis Annual Report 1996 C O N T E N T S Page A Message from Inspector General of Police 1 A Message from Dhaka Metropolitan Police Commissioner 2 SECTION 1 Introduction 3 SECTION 2 Traffic Accident Reporting System 4 Figure 1: Proposed Police Accident Data Units 6 SECTION 3 Analysis of the Data 7 Figure 2: Accidents by Day of Week 8 Figure 3: Accidents by Hour of Day 9 Figure 4: Accidents by Month 10 Figure 5: Accidents by Thana 11 Figure 6: Casualty by Age 13 Figure 7: Casualty by Class of Road User 14 Figure 8: Vehicles Involved and the Injuries of their Drivers 15 Figure 9: Drivers Involved in Accidents 16 Figure 10: Pedestrians Hit by Vehicles 17 Figure 11: Airport Road Dhaka 19 Figure 12: Airport Road-Biswa Road 20 Figure 13: Khilkhet Bus Stand 21 Figure 14: Chittagong Road, Dhaka 22 Figure 15: Vehicles and Casualties on Chittagong Road, Dhaka 24 Figure 16: Jatrabari Crossing 25 SECTION 4 Conclusion 26 SECTION 5 Appendix 27 1. Introduction 1.1 This report was produced for Dhaka Metropolitan Police by Quazi Zakaria Islam of IDC, S. Omar Faruk and M. Khurshid Hossain, Senior Assistant Police Commissioner, DMP HQ from the Institutional Development Component (IDC) of the Second Road Rehabilitation and Maintenance Project (RRMP2). IDC is funded by the British ODA. 1.2 A new traffic accident report form, designed jointly by the Police and IDC, was introduced into the Northern Division of Dhaka Metropolitan Police (DMP) in June 1995. -

Computational Methods
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 + Structural Evolution of LiNn (n=2, 4, 6, 8, and 10) Clusters: Mass Spectrometry and Theoretical Calculations Zhongxue Ge,*,a,b Kewei Ding,a,b Yisu Li,c Hongguang Xu,d Zhaoqiang Chen,c Yiding Ma,b Taoqi Li,b Weiliang Zhu,*,c and Weijun Zheng*,d a State Key Laboratory of Fluorine & Nitrogen Chemicals, Xi’an 710065, China b Xi’an Modern Chemistry Research Institute, Xi’an 710065, China c Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China d Beijing National Laboratory for Molecular Sciences (BNLMS), State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China 1 Fig.S1 Typical mass spectrum of Li-N clusters generated by laser ablation of a LiF target with N2 as carrier gas Fig.S2 Typical mass spectrum of Li-N clusters generated by laser ablation of a LiF:ZrN=4:1 mixture target with N2 as carrier gas Fig.S3 Typical mass spectrum of Li-N clusters generated by laser ablation of a 2 LiF:AlN=2:1 mixture target with N2 as carrier gas Fig.S4 Typical mass spectrum of Li-N clusters generated by laser ablation of a LiF:BN=2:1 mixture target with N2 as carrier gas 3 + Table S1 NBO data for LiN2 (LP: lone pair; BD: bonding; BD*: antibonding;E2: stabilization energy) NPA(Natural Population Analysis) Atom Natural Electron Configuration Natural Charge Li 2s0.012p0.01 0.976 N2 2s1.572p3.573s0.033p0.01 -0.193 -

On the Existence of Partitioned Incomplete Latin Squares with Five Parts
Marshall University Marshall Digital Scholar Mathematics Faculty Research Mathematics 2019 On the Existence of Partitioned Incomplete Latin Squares with Five Parts Jaromy Kuhl Donald McGinn Michael W. Schroeder Follow this and additional works at: https://mds.marshall.edu/mathematics_faculty Part of the Discrete Mathematics and Combinatorics Commons AUSTRALASIAN JOURNAL OF COMBINATORICS Volume 74(1) (2019), Pages 46–60 On the existence of partitioned incomplete Latin squares with five parts Jaromy Kuhl Donald McGinn Department of Mathematics and Statistics University of West Florida Pensacola, FL 32514 U.S.A. [email protected] [email protected] Michael William Schroeder Department of Mathematics Marshall University Huntington, WV 25755 U.S.A. [email protected] Abstract Let a, b, c, d,ande be positive integers. In 1982 Heinrich showed the existence of a partitioned incomplete Latin square (PILS) of type (a, b, c) and (a, b, c, d) if and only if a = b = c and 2a ≥ d. For PILS of type (a, b, c, d, e) with a ≤ b ≤ c ≤ d ≤ e, it is necessary that a + b + c ≥ e, but not sufficient. In this paper we prove an additional necessary condition and classify the existence of PILS of type (a, b, c, d, a + b + c)andPILS with three equal parts. Lastly, we show the existence of a family of PILS in which the parts are nearly the same size. 1 Introduction Let a, n ∈ Z+ and S be a symbol set of order n.Let[n]={1,...,n}, a+S = {a+s | s ∈ S},andaS be the multiset in which each element of S occurs a times. -

Antigenic and Genetic Characteristics of Zoonotic Influenza Viruses and Development of Candidate Vaccine Viruses for Pandemic Preparedness
4 Antigenic and genetic characteristics of zoonotic influenza viruses and development of candidate vaccine viruses for pandemic preparedness February 2019 The development of influenza candidate vaccine viruses (CVVs), coordinated by WHO, remains an essential component of the overall global strategy for pandemic preparedness. Selection and development of CVVs are the first steps towards timely vaccine production and do not imply a recommendation for initiating manufacture. National authorities may consider the use of one or more of these CVVs for pilot lot vaccine production, clinical trials and other pandemic preparedness purposes based on their assessment of public health risk and need. Zoonotic influenza viruses continue to be identified and evolve both genetically and antigenically, leading to the need for additional CVVs for pandemic preparedness purposes. Changes in the genetic and antigenic characteristics of these viruses relative to existing CVVs, and their potential risks to public health justify the need to select and develop new CVVs. This document summarizes the genetic and antigenic characteristics of recent zoonotic influenza viruses and related viruses circulating in animals1 that are relevant to CVV updates. Institutions interested in receiving these CVVs should contact WHO at [email protected] or the institutions listed in announcements published on the WHO website2. Influenza A(H5) Since their emergence in 1997, highly pathogenic avian influenza (HPAI) A(H5) viruses of the A/goose/Guangdong/1/96 haemagglutinin (HA) lineage have become enzootic in some countries, have infected wild birds and continue to cause outbreaks in poultry and sporadic human infections. These viruses have diversified genetically and antigenically, including the emergence of viruses with replacement of the N1 gene segment by N2, N3, N5, N6, N8 or N9 gene segments, leading to the need for multiple CVVs. -

The Regional Dimension a Bayesian Network Analysis
The Regional Dimension A Bayesian Network Analysis Marco Scutari [email protected] Dalle Molle Institute for Artificial Intelligence (IDSIA) December 19, 2019 Overview Bayesian networks: • Network analysis: graphs and arcs. • Arcs and correlation. • Arcs and causality. • Network analysis and linear regression. • Model selection. • Parameter estimation. • Sensitivity analysis. To showcase a proof-of-concept model we will use a sample of SDG indicators from a group of African countries and a group of Asian countries. The General Idea A network analysis is based on the idea that: • quantities of interest can be associated to the nodes of a graph; and that • we can use arcs to represent which variables are correlated with each other. Hence nodes and variables are referred to interchangeably, as well as arcs and correlations (associations more in general). The conceptual steps are: 1. identify the variables that are the quantities of interest and draw one node for each of them; 2. collect data on them, gathering a sample of observations; 3. measure whether they are significantly correlated using the data and draw an arc between each such pair. Data, Variables and Networks 3. measure correlations and draw the arcs. 1. identify the variables; N1 N2 N3 N4 N5 N6 N2 2. collect data; N1 N2 N3 N4 N5 N6 N5 N6 0.11 0.67 0.37 1.56 -0.77 -0.77 -0.19 -0.52 0.88 -0.44 -0.38 1.28 -1.34 0.19 -1.26 -0.24 -0.31 -0.16 -1.23 -1.01 0.13 -1.15 0.22 0.34 0.85 0.75 0.71 1.17 -0.70 0.29 N3 ⋮ ⋮ ⋮ ⋮ ⋮ ⋮ N1 N4 Arcs and Correlations How do we interpret arcs? • Nodes that are directly connected with each other are directly correlated: changes in one node suggest changes in the nodes that are directly connected with it.