Spatial Modelling of Bacterial Diversity Over the Selected Regions in Bangladesh by Next-Generation Sequencing: Role of Water Temperature
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

12. FORMULATION of the URBAN TRANSPORT MASTER PLAN Development of the RSTP Urban Transportation Master Plan (1) Methodology
The Project on The Revision and Updating of the Strategic Transport Plan for Dhaka (RSTP) Final Report 12. FORMULATION OF THE URBAN TRANSPORT MASTER PLAN Development of the RSTP Urban Transportation Master Plan (1) Methodology The development of the RSTP Urban Transportation Master Plan adopted the following methodology (see Figure 12.1): (i) Elaborate the master plan network through a screen line analysis by comparing the network capacity and future demand. (ii) Identify necessary projects to meet future demand at the same time avoiding excessive capacity. (iii) Conducts economic evaluation of each project to give priority on projects with higher economic return. (iv) Conduct preliminary environmental assessment of every project and consider countermeasures against environmental problems, if any. (v) Make a final prioritization of all physical projects by examining their respective characteristics from different perspectives. (vi) Classify the projects into three categories, namely short-, medium- and long-term projects, by considering the financial constraints. (vii) Prepare an action plan for short-term projects together with “soft” measures. Mid-term Project Source: RSTP Study Team Figure 12.1 Development Procedure for the Master Plan 12-1 The Project on The Revision and Updating of the Strategic Transport Plan for Dhaka (RSTP) Final Report (2) Output of the Transportation Network Plan The RSTP urban transportation network plan was developed based on a review and a modification of the STP network plan. The main points of the modification or adoption of the STP network master plan are as follows: i. Harmonization with future urban structure, land-use plan and development of network plan. -

Effect of Nitrogen and Boron on the Yield of Wheat Cv. Kanchan
J. Bangladesh Agril. Univ. 3(2): 215-220, 2005 ISSN 1810-3030 Effect of nitrogen and boron on the yield of wheat cv. Kanchan M.M. Khan', A.K. Hasan2, M.H. Rashid2, and F. Ahrned3 Palli Karma Sahayak Foundation (PKSF), Sher-e-Banglanagar, Dhaka, 2Department of Agronomy and 3 Department of Agroforestry, Bangladesh Agricultural University, Mymensingh Abstract An experiment was carried out at the Agronomy Field Laboratory, Bangladesh Agricultural University, Mymensingh during the period from January to April 2004 to study the effect of different levels of nitrogen and boron on the yield of wheat cv. Kanchan. The treatments included four levels of nitrogen VIZ., 45, 60, 85 and 110 kg N ha-1 and four levels of boron viz., 0, 1, 2 and 3 kg B ha-1. The experiment was laid out in a Randomized Complete Block Design with three replications. The results revealed that Yield and yield contributing characters were influenced significantly by both levels of nitrogen and boron. Among the levels of nitrogen, 110 kg N ha-1 produced the highest grain (5.54 t ha-1)and straw (8.21 t ha-1 ) yields. The lowest grain (3.23 t ha-1) and straw (5.52 t ha-1) yields were observed with the application of 45 kg N ha-1. The highest grain (4.95 t ha-1)and straw (7.38 t ha 1) yields were produced With 3 kg B ha-1. The minimum grain (3.59 t ha-1)and straw (6.14 t ha-1)yields were found in the control treatment. -

Connecting Bangladesh: Economic Corridor Network
Connecting Bangladesh: Economic Corridor Network Economic corridors are anchored on transport corridors, and international experience suggests that the higher the level of connectivity within and across countries, the higher the level of economic growth. In this paper, a new set of corridors is being proposed for Bangladesh—a nine-corridor comprehensive integrated multimodal economic corridor network resembling the London Tube map. This paper presents the initial results of the research undertaken as an early step of that development effort. It recommends an integrated approach to developing economic corridors in Bangladesh that would provide a strong economic foundation for the construction of world-class infrastructure that, in turn, could support the growth of local enterprises and attract foreign investment. About the Asian Development Bank COnnecTING BANGLADESH: ADB’s vision is an Asia and Pacific region free of poverty. Its mission is to help its developing member countries reduce poverty and improve the quality of life of their people. Despite the region’s many successes, it remains home to a large share of the world’s poor. ADB is committed to reducing poverty through inclusive economic growth, environmentally sustainable growth, and regional integration. ECONOMIC CORRIDOR Based in Manila, ADB is owned by 67 members, including 48 from the region. Its main instruments for helping its developing member countries are policy dialogue, loans, equity investments, guarantees, grants, NETWORK and technical assistance. Mohuiddin Alamgir -

Table S4. Phylogenetic Distribution of Bacterial and Archaea Genomes in Groups A, B, C, D, and X
Table S4. Phylogenetic distribution of bacterial and archaea genomes in groups A, B, C, D, and X. Group A a: Total number of genomes in the taxon b: Number of group A genomes in the taxon c: Percentage of group A genomes in the taxon a b c cellular organisms 5007 2974 59.4 |__ Bacteria 4769 2935 61.5 | |__ Proteobacteria 1854 1570 84.7 | | |__ Gammaproteobacteria 711 631 88.7 | | | |__ Enterobacterales 112 97 86.6 | | | | |__ Enterobacteriaceae 41 32 78.0 | | | | | |__ unclassified Enterobacteriaceae 13 7 53.8 | | | | |__ Erwiniaceae 30 28 93.3 | | | | | |__ Erwinia 10 10 100.0 | | | | | |__ Buchnera 8 8 100.0 | | | | | | |__ Buchnera aphidicola 8 8 100.0 | | | | | |__ Pantoea 8 8 100.0 | | | | |__ Yersiniaceae 14 14 100.0 | | | | | |__ Serratia 8 8 100.0 | | | | |__ Morganellaceae 13 10 76.9 | | | | |__ Pectobacteriaceae 8 8 100.0 | | | |__ Alteromonadales 94 94 100.0 | | | | |__ Alteromonadaceae 34 34 100.0 | | | | | |__ Marinobacter 12 12 100.0 | | | | |__ Shewanellaceae 17 17 100.0 | | | | | |__ Shewanella 17 17 100.0 | | | | |__ Pseudoalteromonadaceae 16 16 100.0 | | | | | |__ Pseudoalteromonas 15 15 100.0 | | | | |__ Idiomarinaceae 9 9 100.0 | | | | | |__ Idiomarina 9 9 100.0 | | | | |__ Colwelliaceae 6 6 100.0 | | | |__ Pseudomonadales 81 81 100.0 | | | | |__ Moraxellaceae 41 41 100.0 | | | | | |__ Acinetobacter 25 25 100.0 | | | | | |__ Psychrobacter 8 8 100.0 | | | | | |__ Moraxella 6 6 100.0 | | | | |__ Pseudomonadaceae 40 40 100.0 | | | | | |__ Pseudomonas 38 38 100.0 | | | |__ Oceanospirillales 73 72 98.6 | | | | |__ Oceanospirillaceae -

Diversity of Biodeteriorative Bacterial and Fungal Consortia in Winter and Summer on Historical Sandstone of the Northern Pergol
applied sciences Article Diversity of Biodeteriorative Bacterial and Fungal Consortia in Winter and Summer on Historical Sandstone of the Northern Pergola, Museum of King John III’s Palace at Wilanow, Poland Magdalena Dyda 1,2,* , Agnieszka Laudy 3, Przemyslaw Decewicz 4 , Krzysztof Romaniuk 4, Martyna Ciezkowska 4, Anna Szajewska 5 , Danuta Solecka 6, Lukasz Dziewit 4 , Lukasz Drewniak 4 and Aleksandra Skłodowska 1 1 Department of Geomicrobiology, Institute of Microbiology, Faculty of Biology, University of Warsaw, Miecznikowa 1, 02-096 Warsaw, Poland; [email protected] 2 Research and Development for Life Sciences Ltd. (RDLS Ltd.), Miecznikowa 1/5a, 02-096 Warsaw, Poland 3 Laboratory of Environmental Analysis, Museum of King John III’s Palace at Wilanow, Stanislawa Kostki Potockiego 10/16, 02-958 Warsaw, Poland; [email protected] 4 Department of Environmental Microbiology and Biotechnology, Institute of Microbiology, Faculty of Biology, University of Warsaw, Miecznikowa 1, 02-096 Warsaw, Poland; [email protected] (P.D.); [email protected] (K.R.); [email protected] (M.C.); [email protected] (L.D.); [email protected] (L.D.) 5 The Main School of Fire Service, Slowackiego 52/54, 01-629 Warsaw, Poland; [email protected] 6 Department of Plant Molecular Ecophysiology, Institute of Experimental Plant Biology and Biotechnology, Faculty of Biology, University of Warsaw, Miecznikowa 1, 02-096 Warsaw, Poland; [email protected] * Correspondence: [email protected] or [email protected]; Tel.: +48-786-28-44-96 Citation: Dyda, M.; Laudy, A.; Abstract: The aim of the presented investigation was to describe seasonal changes of microbial com- Decewicz, P.; Romaniuk, K.; munity composition in situ in different biocenoses on historical sandstone of the Northern Pergola in Ciezkowska, M.; Szajewska, A.; the Museum of King John III’s Palace at Wilanow (Poland). -
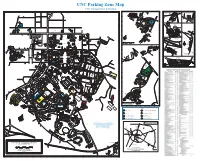
UNC Parking Zone Map UNC Transportation & Parking
UNC Parking Zone Map UNC Transportation & Parking Q R S T U V W X Y Z A B C D E F G H I J K L M N O P 26 **UNC LEASES SPACE CAROLINA . ROAD IN THESE BUILDINGS 21 21 MT HOMESTEAD NORTH LAND MGMT. PINEY OPERATIONS CTR. VD. (NC OFFICE HORACE WILLIAMS AIRPORT VD., HILL , JR. BL “RR” 41 1 1 Resident 41 CommuterRR Lot R12 UNC VD AND CHAPEL (XEROX) TE 40 MLK BL A PRINTING RIVE EXTENSION MLK BL ESTES D SERVICES TIN LUTHER KING TERST PLANT N O I AHEC T EHS HOMESTEAD ROAD MAR HANGER VD. 86) O I-40 STORAGE T R11 TH (SEE OTHER MAPS) 22 22 O 720, 725, & 730 MLK, JR. BL R1 T PHYSICAL NOR NORTH STREET ENVRNMEN HL .3 MILES TO TH. & SAFETY ESTES DRIVE 42 COMMUTER LOT T. 42 ER NC86 ELECTRICAL DISTRICENTBUTION OPERATIONS SURPLUS WA REHOUSE N1 ST GENERAL OREROOM 2 23 23 2 R1 CHAPEL HILL ES MLK JR. BOULE NORTH R1 ARKING ARD ILITI R1 / R2OVERFLOW ZONEP V VICES C R A F SHOPS GY SE EY 43 RN 43 ENERBUILDING CONSTRUCTION PRITCHARD STREET R1 NC 86 CHURCH STREET . HO , JR. BOULE ES F R1 / V STREET SER L BUILDING VICE ARD A ST ATIO GI EET N TR AIRPOR R2 S T DRIVE IN LUTHER KING BRANCH T L MAR HIL TH WEST ROSEMARY STREET EAST ROSEMARY STREET L R ACILITIES DRIVE F A NO 24 STUDRT 24 TH COLUMBI IO CHAPE R ADMINIST OFF R NO BUILDINGICE ATIVE R10 1700 N9 MLK 208 WEST 3 N10 FRANKLIN ST. -

Microbial Community Structure in Rice, Crops, and Pastures Rotation Systems with Different Intensification Levels in the Temperate Region of Uruguay
Supplementary Material Microbial community structure in rice, crops, and pastures rotation systems with different intensification levels in the temperate region of Uruguay Sebastián Martínez Table S1. Relative abundance of the 20 most abundant bacterial taxa of classified sequences. Relative Taxa Phylum abundance 4,90 _Bacillus Firmicutes 3,21 _Bacillus aryabhattai Firmicutes 2,76 _uncultured Prosthecobacter sp. Verrucomicrobia 2,75 _uncultured Conexibacteraceae bacterium Actinobacteria 2,64 _uncultured Conexibacter sp. Actinobacteria 2,14 _Nocardioides sp. Actinobacteria 2,13 _Acidothermus Actinobacteria 1,50 _Bradyrhizobium Proteobacteria 1,23 _Bacillus Firmicutes 1,10 _Pseudolabrys_uncultured bacterium Proteobacteria 1,03 _Bacillus Firmicutes 1,02 _Nocardioidaceae Actinobacteria 0,99 _Candidatus Solibacter Acidobacteria 0,97 _uncultured Sphingomonadaceae bacterium Proteobacteria 0,94 _Streptomyces Actinobacteria 0,91 _Terrabacter_uncultured bacterium Actinobacteria 0,81 _Mycobacterium Actinobacteria 0,81 _uncultured Rubrobacteria Actinobacteria 0,77 _Xanthobacteraceae_uncultured forest soil bacterium Proteobacteria 0,76 _Streptomyces Actinobacteria Table S2. Relative abundance of the 20 most abundant fungal taxa of classified sequences. Relative Taxa Orden abundance. 20,99 _Fusarium oxysporum Ascomycota 11,97 _Aspergillaceae Ascomycota 11,14 _Chaetomium globosum Ascomycota 10,03 _Fungi 5,40 _Cucurbitariaceae; uncultured fungus Ascomycota 5,29 _Talaromyces purpureogenus Ascomycota 3,87 _Neophaeosphaeria; uncultured fungus Ascomycota -

Monitoring of Biofouling Communities in a Portuguese Port Using a Combined Morphological and Metabarcoding Approach Joana Azevedo 1,2,3, Jorge T
www.nature.com/scientificreports OPEN Monitoring of biofouling communities in a Portuguese port using a combined morphological and metabarcoding approach Joana Azevedo 1,2,3, Jorge T. Antunes1,2,3, André M. Machado 1, Vitor Vasconcelos1,2, Pedro N. Leão 1* & Elsa Froufe 1* Marine biofouling remains an unsolved problem with a serious economic impact on several marine associated industries and constitutes a major vector for the spread of non-indigenous species (NIS). The implementation of biofouling monitoring programs allows for better fouling management and also for the early identifcation of NIS. However, few monitoring studies have used recent methods, such as metabarcoding, that can signifcantly enhance the detection of those species. Here, we employed monthly monitoring of biofouling growth on stainless steel plates in the Atlantic Port of Leixões (Northern Portugal), over one year to test the efect of commercial anti-corrosion paint in the communities. Fouling organisms were identifed by combining morpho-taxonomy identifcation with community DNA metabarcoding using multiple markers (16S rRNA, 18S rRNA, 23S rRNA, and COI genes). The dominant colonizers found at this location were hard foulers, namely barnacles and mussels, while other groups of organisms such as cnidarians, bryozoans, and ascidians were also abundant. Regarding the temporal dynamics of the fouling communities, there was a progressive increase in the colonization of cyanobacteria, green algae, and red algae during the sampled period with the replacement of less abundant groups. The tested anticorrosion paint demonstrated to have a signifcant prevention efect against the biofouling community resulting in a biomass reduction. Our study also reports, for the frst time, 29 NIS in this port, substantiating the need for the implementation of recurring biofouling monitoring programs in ports and harbours. -

Systema Naturae 2000 (Phylum, 6 Nov 2017)
The Taxonomicon Systema Naturae 2000 Classification of Domain Bacteria (prokaryotes) down to Phylum Compiled by Drs. S.J. Brands Universal Taxonomic Services 6 Nov 2017 Systema Naturae 2000 - Domain Bacteria - Domain Bacteria Woese et al. 1990 1 Genus †Eoleptonema Schopf 1983, incertae sedis 2 Genus †Primaevifilum Schopf 1983, incertae sedis 3 Genus †Archaeotrichion Schopf 1968, incertae sedis 4 Genus †Siphonophycus Schopf 1968, incertae sedis 5 Genus Bactoderma Tepper and Korshunova 1973 (Approved Lists 1980), incertae sedis 6 Genus Stibiobacter Lyalikova 1974 (Approved Lists 1980), incertae sedis 7.1.1.1.1.1 Superphylum "Proteobacteria" Craig et al. 2010 1.1 Phylum "Alphaproteobacteria" 1.2.1 Phylum "Acidithiobacillia" 1.2.2.1 Phylum "Gammaproteobacteria" 1.2.2.2.1 Candidate phylum Muproteobacteria (RIF23) Anantharaman et al. 2016 1.2.2.2.2 Phylum "Betaproteobacteria" 2 Phylum "Zetaproteobacteria" 7.1.1.1.1.2 Phylum "Deltaproteobacteria_1" 7.1.1.1.2.1.1.1 Phylum "Deltaproteobacteria" [polyphyletic] 7.1.1.1.2.1.1.2.1 Phylum "Deltaproteobacteria_2" 7.1.1.1.2.1.1.2.2 Phylum "Deltaproteobacteria_3" 7.1.1.1.2.1.2 Candidate phylum Dadabacteria (CSP1-2) Hug et al. 2015 7.1.1.1.2.2.1 Candidate phylum "MBNT15" 7.1.1.1.2.2.2 Candidate phylum "Uncultured Bacterial Phylum 10 (UBP10)" Parks et al. 2017 7.1.1.2.1 Phylum "Nitrospirae_1" 7.1.1.2.2 Phylum Chrysiogenetes Garrity and Holt 2001 7.1.2.1.1 Phylum "Nitrospirae" Garrity and Holt 2001 [polyphyletic] 7.1.2.1.2.1.1 Candidate phylum Rokubacteria (CSP1-6) Hug et al. -

Marine Sediments Illuminate Chlamydiae Diversity and Evolution
Supplementary Information for: Marine sediments illuminate Chlamydiae diversity and evolution Jennah E. Dharamshi1, Daniel Tamarit1†, Laura Eme1†, Courtney Stairs1, Joran Martijn1, Felix Homa1, Steffen L. Jørgensen2, Anja Spang1,3, Thijs J. G. Ettema1,4* 1 Department of Cell and Molecular Biology, Science for Life Laboratory, Uppsala University, SE-75123 Uppsala, Sweden 2 Department of Earth Science, Centre for Deep Sea Research, University of Bergen, N-5020 Bergen, Norway 3 Department of Marine Microbiology and Biogeochemistry, NIOZ Royal Netherlands Institute for Sea Research, and Utrecht University, NL-1790 AB Den Burg, The Netherlands 4 Laboratory of Microbiology, Department of Agrotechnology and Food Sciences, Wageningen University, 6708 WE Wageningen, The Netherlands. † These authors contributed equally * Correspondence to: Thijs J. G. Ettema, Email: [email protected] Supplementary Information Supplementary Discussions ............................................................................................................................ 3 1. Evolutionary relationships within the Chlamydiae phylum ............................................................................. 3 2. Insights into the evolution of pathogenicity in Chlamydiaceae ...................................................................... 8 3. Secretion systems and flagella in Chlamydiae .............................................................................................. 13 4. Phylogenetic diversity of chlamydial nucleotide transporters. .................................................................... -

Rumen Bacterial Community of Young and Adult of Reindeer
Open Agriculture. 2020; 5: 10-20 Research Article Kasim A. Laishev, Larisa A. Ilina, Valentina A. Filippova, Timur P. Dunyashev, Georgiy Yu. Laptev, Evgeny V. Abakumov* Rumen bacterial community of young and adult of reindeer (rangifer tarandus) from Yamalo-Nenets Autonomous District of Russia https://doi.org/10.1515/opag-2020-0001 that the presence of the phylum Verrucomicrobia, and the received June 15, 2019; accepted December 3, 2019 genera Stenotrophomonas, Pseudomonas, etc., may be specific to Nenets breed reindeer and have a pattern with Abstract: The aim of the work was to compare the taxo- their presence on various plants and lichens that are part nomic composition of the rumen procariotic community of the reindeer diet. This is partially confirmed by data on in young and adult individuals of Nenets breed rein- plants microbiome taxonomy. deer (Rangifer tarandus ) from the central part of the Yamal region by using the NGS method (next generation Keywords: Reindeer; Rumen; Microbiome; Polar environ- sequencing) and compare the microbiome composition ments; Vegetation materials; Metagenomics; taxonomy of reindeer with the microbiome of their initial vegetation food material. The obtained data showed that the domi- nant position in microbial communities, like that of other ruminants, was occupied by representatives of phylum 1 Introduction Firmicutes and Bacteroidetes, whose total share between observed groups did not differ significantly. The compo- Currently, agriculture in the Russian Arctic is an inten- sition of the microbiome of the rumen of the investigated sively developing part of the local economic. Strong group of animals was completely different from the micro- localization of industrial and agricultural activity in biome structure of the initial vegetation cover. -
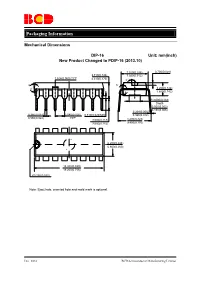
Mechanical-Dimensions-BCD.Pdf
Packaging Information Mechanical Dimensions DIP-16 Unit: mm(inch) New Product Changed to PDIP-16 (2013.10) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060) TYP 4.310(0.170) 5° 6° 6° 4° 3.200(0.126) 4° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014) 0.560(0.022) TYP 3.000(0.118) 8.200(0.323) 3.600(0.142) 9.400(0.370) 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optional. Dec. 2014 BCD Semiconductor Manufacturing Limited Packaging Information Mechanical Dimensions DIP-16 (Special for AZ4052) Unit: mm(inch) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060)T YP 4.310(0.170) 5 ° 6 ° 6 ° 4 ° 3.200(0.126) 4 ° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014 ) 0.560(0.022) TYP 3.000(0.118) 8.500(0.335) 3.600(0.142) TYP 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optiona.l Dec.