Estimating the Burden of Α-Thalassaemia in Thailand Using A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
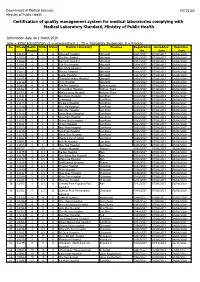
Certification of Quality Management System for Medical Laboratories Complying with Medical Laboratory Standard, Ministry of Public Health
Department of Medical Sciences F0715102 Ministry of Public Health Certification of quality management system for medical laboratories complying with Medical Laboratory Standard, Ministry of Public Health Information date on 1 March 2019 new = initial accreditation, r1 = reassessment 1 , TS = Temporary Suspension, P = Process No. HCode Health RMSc Status Medical Laboratory Province Registration Accredited Expiration Region No. Date Date 1 10673 2 2 r1 Uttaradit Hospital Uttaradit 0001/2557 07/08/2017 06/08/2020 2 11159 2 2 r1 Tha Pla Hospital Uttaradit 0002/2557 07/08/2017 06/08/2020 3 11160 2 2 r1 Nam Pat Hospital Uttaradit 0003/2557 07/08/2017 06/08/2020 4 11161 2 2 r1 Fak Tha Hospital Uttaradit 0004/2557 07/08/2017 06/08/2020 5 11162 2 2 r1 Ban Khok Hospital Uttaradit 0005/2557 07/08/2017 06/08/2020 6 11163 2 2 r1 Phichai Hospital Uttaradit 0006/2557 07/08/2017 06/08/2020 7 11164 2 2 r1 Laplae Hospital Uttaradit 0007/2557 07/08/2017 06/08/2020 8 11165 2 2 r1 ThongSaenKhan Hospital Uttaradit 0008/2557 07/08/2017 06/08/2020 9 11158 2 2 r1 Tron Hospital Uttaradit 0009/2557 07/08/2017 06/08/2020 10 10863 4 4 r1 Pak Phli Hospital Nakhonnayok 0010/2557 07/08/2017 06/08/2020 11 10762 4 4 r1 Thanyaburi Hospital Pathum Thani 0011/2557 07/08/2017 06/08/2020 12 10761 4 4 r1 Klong Luang Hospital Pathum Thani 0012/2557 07/08/2017 06/08/2020 13 11141 1 1 P Ban Hong Hospital LamPhun 0014/2557 07/08/2014 06/08/2017 14 11142 1 1 P Li Hospital LamPhun 0015/2557 07/08/2014 06/08/2017 15 11144 1 1 P Pa Sang Hospital LamPhun 0016/2557 07/08/2014 06/08/2017 -

Clinical Epidemiology of 7126 Melioidosis Patients in Thailand and the Implications for a National Notifiable Diseases Surveilla
applyparastyle “fig//caption/p[1]” parastyle “FigCapt” View metadata, citation and similar papers at core.ac.uk brought to you by CORE Open Forum Infectious Diseases provided by Apollo MAJOR ARTICLE Clinical Epidemiology of 7126 Melioidosis Patients in Thailand and the Implications for a National Notifiable Diseases Surveillance System Viriya Hantrakun,1, Somkid Kongyu,2 Preeyarach Klaytong,1 Sittikorn Rongsumlee,1 Nicholas P. J. Day,1,3 Sharon J. Peacock,4 Soawapak Hinjoy,2,5 and Direk Limmathurotsakul1,3,6, 1Mahidol-Oxford Tropical Medicine Research Unit (MORU), Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand, 2 Epidemiology Division, Department of Disease Control, Ministry of Public Health, Nonthaburi, Thailand, 3 Centre for Tropical Medicine and Global Health, Nuffield Department of Clinical Medicine, Old Road Campus, University of Oxford, Oxford, United Kingdom, 4 Department of Medicine, University of Cambridge, Cambridge, United Kingdom, 5 Office of International Cooperation, Department of Disease Control, Ministry of Public Health, Nonthaburi, Thailand, and 6 Department of Tropical Hygiene, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand Background. National notifiable diseases surveillance system (NNDSS) data in developing countries are usually incomplete, yet the total number of fatal cases reported is commonly used in national priority-setting. Melioidosis, an infectious disease caused by Burkholderia pseudomallei, is largely underrecognized by policy-makers due to the underreporting of fatal cases via the NNDSS. Methods. Collaborating with the Epidemiology Division (ED), Ministry of Public Health (MoPH), we conducted a retrospec- tive study to determine the incidence and mortality of melioidosis cases already identified by clinical microbiology laboratories nationwide. A case of melioidosis was defined as a patient with any clinical specimen culture positive for B. -

Mortality Risk and Temporal Patterns of Atrial Fibrillation in the Nationwide
medRxiv preprint doi: https://doi.org/10.1101/2021.01.30.21250715; this version posted June 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Mortality Risk and Temporal Patterns of Atrial Fibrillation in the Nationwide Registry Apiyasawat Short Title: Mortality Risk in Non-paroxysmal AF Sirin Apiyasawat, MD1; Sakaorat Kornbongkotmas, MD2; Ply Chichareon, MD3; Rungroj Krittayaphong, MD4; for the COOL-AF Investigators Links to Affiliations: 1 Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand 2 Queen Savang Vadhana Memorial Hospital, Chonburi, Thailand 3 Faculty of Medicine, Prince of Songkla University, Songkla, Thailand 4 Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand Address for correspondence: Rungroj Krittayaphong, MD Division of Cardiology, Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand Phone: (66) 2-419-6104; Fax: (66) 2-412-7412, E-mail: [email protected] Word Count: 2299 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2021.01.30.21250715; this version posted June 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . -

Saturday 5 September 2015
SATURDAY 5 SEPTEMBER 2015 SATURDAY 5 SEPTEMBER Registration Desk 0745-1730 Registration Desk, SECC for Pre-conference Workshop and Course Participants Location: Hall 4, SECC Group Meeting 1000-1700 AMEE Executive Committee Meeting (Closed Meeting) Location: Green Room 10, Back of Hall 4 AMEE-Essential Skills in Medical Education (ESME) Courses Pre-registration is essential and lunch will be provided. 0830-1700 ESME – Essential Skills in Medical Education Location: Argyll I, Crowne Plaza 0845-1630 ESMEA – Essential Skills in Medical Education Assessment Location: Argyll III, Crowne Plaza 0845-1630 RESME – Research Essential Skills in Medical Education Location: Argyll II, Crowne Plaza 0845-1700 ESMESim - Essential Skills in Simulation-based Healthcare Instruction Location: Castle II, Crowne Plaza 0900-1700 ESCEPD – Essential Skills in Continuing Education and Professional Development Location: Castle 1, Crowne Plaza 1000-1330 ESCEL – Essential Skills in Computer-Enhanced Learning Location: Carron 2, SECC Course Pre-registration is essential and lunch will be provided. 0830-1630 ASME-FLAME - Fundamentals of Leadership and Management in Education Location: Castle III, Crowne Plaza Masterclass Pre-registration is essential and lunch will be provided. 0915-1630 MC1 Communication Matters: Demystifying simulation design, debriefing and facilitation practice Kerry Knickle (Standardized Patient Program, Faculty of Medicine, University of Toronto, Canada); Nancy McNaughton, Diana Tabak) University of Toronto, Centre for Research in Education, Standardized -

Age-Related Clinical Outcomes of Patients with Non-Valvular Atrial Fibrillation: Insights from the COOL-AF Registry
Clinical Interventions in Aging Dovepress open access to scientific and medical research Open Access Full Text Article ORIGINAL RESEARCH Age-Related Clinical Outcomes of Patients with Non-Valvular Atrial Fibrillation: Insights from the COOL-AF Registry Rungroj Krittayaphong1 Purpose: We aimed to compare the rate of clinical outcomes among three age groups (<65, Thanita Boonyapiphat2 65–74, and ≥75 years) of adult patients with non-valvular atrial fibrillation (NVAF). Chaiyasith Wongvipaporn3 Patients and Methods: We prospectively enrolled NVAF patients from 27 Thailand Poom Sairat1 medical centers. The following were collected at baseline: demographic data, risk factors, comorbid conditions, laboratory data, and medications. The clinical outcomes were ischemic On behalf of the COOL-AF stroke (IS) or transient ischemic attack (TIA), major bleeding (MB), intracerebral hemor Investigators rhage (ICH), heart failure (HF), and death. All events were adjudicated. Patients were categorized according to age group into three groups; age <65, 65–74, and ≥75 years. 1Division of Cardiology, Department of Results: Among the 3402 patients that were enrolled during 2014–2017, the mean age was 67.4 Medicine, Faculty of Medicine Siriraj ±11.3 years, and 2073 (60.9%) were older. The average follow-up was 25.7±10.6 months. Oral Hospital, Mahidol University, Bangkok, Thailand; 2Division of Cardiology, anticoagulants were given in 75.4% of patients (91.1% of OAC was warfarin). The incidence rate Department of Medicine, Lampang of IS/TIA, MB, ICH, HF, and death was 1.43 (1.17–1.74), 2.11 (1.79–2.48), 0.70 (0.52–0.92), Hospital, Lampang, Thailand; 3Division of Cardiology, Department of Medicine, 3.03 (2.64–3.46), and 3.77 (3.33–4.24) per 100 person-years, respectively. -

Chapter 6 the Expansion New Membership Recruitment Area of Thai Maternal and Child Health Network Under the Royal Patronage 6
Chapter 6 The Expansion New Membership Recruitment Area of Thai Maternal and Child Health Network under the Royal Patronage 6 Thrathip Kolatat, Chantima Charastong At present, Thai Maternal and Child Health Network Board of Committee under the Royal Patronage has established project purposes that meet the principle objective, which is to lower the rate of preterm births. However, the board’s reexamination of the issue reveals the aforementioned strategy can be elevated to be policy-level strategy, the process of which includes setting up the clear strategic targets and public services, as well as considering the differences between service areas. It has also been suggested that personnel in those areas should be the ones coming up with action plans, to successfully reach the ultimate outcome1. Having studied the fundamental patterns of Thai Maternal and Child Health Network’s project management according to national context, the board has established an expansion model, which foresees the project expanding into various other areas, being carried out in a direction towards the expected outcome. Establishing a strategy map in each area begins with strengthening work forces in the provincial level. Since the public health system is directed by Office of Provincial Chief Medical Officer, which provides management and supports to community and district health promotion hospitals, not the general and regional hospitals. Thus, if there is to be an integration of both health promotion and treatment, an operational conference is essential. The conference would allow idea sharing and discussion between the multidisciplinary involved, namely obstetricians, pediatricians, general practitioners, registered nurses (from prenatal clinics, delivery rooms, emergency rooms, neonatal intensive care unit, neonatal wards and follow-up clinics, etc.), as well as public health technical officers, social medicine officers from community hospitals, general hospitals and regional hospitals. -

Outcome Disparities by Insurance Plan and Educational Attainment In
medRxiv preprint doi: https://doi.org/10.1101/2021.04.25.21256068; this version posted April 28, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Outcome Disparities by Insurance Plan and Educational Attainment in Patients with Atrial Fibrillation Apiyasawat Short Title: Education Disparities and AF Outcomes Sirin Apiyasawat, MD1; Tomon Thongsri, MD2, Kulyot Jongpiputvanich, MD3, Rungroj Krittayaphong, MD4; for the COOL-AF Investigators 1 Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand 2 Buddhachinaraj Hospital, Phitsanulok, Thailand 3 Faculty of Medicine, Vajira Hospital, Navamindradhiraj University, Bangkok, Thailand 4 Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand Address for correspondence: Rungroj Krittayaphong, MD Division of Cardiology, Department of Medicine, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand Phone: (66) 2-419-6104; Fax: (66) 2-412-7412, E-mail: [email protected] Word Counts: 2990 Keywords: atrial fibrillation, health insurance, education, mortality, registry Apiyasawat,NOTE: This preprint et al. reports new research that has not been certified1 by peer review Educationand should not Disparities be used to guide and clinical AF Outcomes practice. medRxiv preprint doi: https://doi.org/10.1101/2021.04.25.21256068; this version posted April 28, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. -

วารสารวิจัยและนวัตกรรมทางสุขภาพ Journal of Health Research and Innovation
วิทยาลัยพยาบาลบรมราชชนนี สุราษฎร์ธานี The Conducting of Self-Help Group in Adolescents with Cancer: Nurses’ Roles วารสารวิจัยและนวัตกรรมทางสุขภาพ Journal of Health Research and Innovation ปี ที ่ 3 ฉบับที ่ 2 กรกฎาคม – ธันวาคม 2563 วัตถุประสงค์ 1. เพื่อเผยแพร่ผลงานวิจัย บทความทางวิชาการและนวัตกรรมทางสุขภาพของอาจารย์ บุคลากร นักศึกษา วิทยาลัยพยาบาลบรมราชชนนี สุราษฎร์ธานี ในด้านการแพทย์ การพยาบาล การสาธารณสุข การศึกษาใน สาขาวิทยาศาสตร์สุขภาพ และด้านอื่น ๆ ที่เกี่ยวข้องกับวิทยาศาสตร์สุขภาพ 2. เพื่อเผยแพร่ผลงานวิจัย บทความทางวิชาการและนวัตกรรมทางสุขภาพของบุคลากรทางการแพทย์ นักวิชาการผู้ปฏิบัติงานในศาสตร์ที่เกี่ยวข้องตลอดจนศิษย์เก่า และผู้สนใจ ในด้านการแพทย์ การพยาบาล การสาธารณสุข การศึกษาในสาขาวิทยาศาสตร์สุขภาพ และด้านอื่น ๆ ที่เกี่ยวข้องกับวิทยาศาสตร์สุขภาพ 3. เพื่อสร้างเครือข่ายทางวิชาการทั้งในวิทยาลัยพยาบาลบรมราชชนนี สุราษฎร์ธานี และสถาบันวิชาชีพ ที่เกี่ยวข้อง 4. เพื่อตอบสนองพันธกิจหลักในการสร้างองค์ความรู้และการเผยแพร่ผลงานวิชาการและงานวิจัยของ วิทยาลัยพยาบาลบรมราชชนนี สุราษฎร์ธานี สำนักงำน บรรณาธิการวารสาร วิทยาลัยพยาบาลบรมราชชนนี สุราษฎร์ธานี 56/6 หมู่ 2 ถ.ศรีวิชัย ตาบล มะขามเตี้ย อาเภอเมือง จังหวัดสุราษฎร์ธานี 84000 โทร. 0-7728-7816 ต่อ 218 โทรสาร 0-7727-2571 http://www.bcnsurat.ac.th E-mail: [email protected] พมิ พท์ ี่ โรงพิมพ์เลิศไชย 16/4-6 ถนนไตรอนสุ นธิ์ ตาบลตลาด อาเภอเมืองสุราษฎร์ธานี จังหวัดสุราษฎร์ธานี โทรศัพท์ 0 7727 3973 โทรสาร 0 7729 9521 วารสารวจิ ยั และนวตั กรรมทางสขุ ภาพ เป็นวารสารทมี่ ผี ูท้ รงคณุ วฒุ ติ รวจสอบเนอื้ หาบทความเพอื่ ลงตพี มิ พจ์ า นวน 2 ท่านต่อบทความ และ บทความหรอื ขอ้ คดิ เหน็ ใด ๆ ทปี่ รากฏในวารสารวจิ -

The Thai Journal of SURGERY
ISSN 0125-6068 E-ISSN 2697-5858 The Thai Journal of SURGERY Official Publication of The Royal College of Surgeons of Thailand www.tci-thaijo.org/index.php/ThaiJSurg/index Volume 41 July-September 2020 Number 3 REVIEW ARTICLE 65 Nipple Sparing Mastectomy in Breast Cancer Patients Chayanoot Rattadilok, Prakasit Chirappapha, Prapa Rattadilok ORIGINAL ARTICLE 79 Simplified ERAS Program for Elective Colorectal Surgery at Suratthani Hospital Sorawat Janwanitchsthaporn, Jirapong Intarasompun CASE REPORTS 84 A Report of Three Patients Surviving Near-Fatal Blunt Cardiac Rupture Komkrit Komuttarin, Nisit Poolthananant, Araya Thitisurawat, Witchayoot Teerapinyo 92 Klebsiella Distinctive Syndrome Presenting with Muscular Abscess and Osteomyelitis Nutthawut Akaranuchat, Chanon Parkpinyo ABSTRACTS 96 Abstracts of the 45th Annual Scientific Congress of The Royal College of Surgeons of Thailand, 8-11 October 2020, RCST Virtual Annual Congress 2020 (Part I) Secretariat Office : Royal Golden Jubilee Building, 2 Soi Soonvijai, New Petchaburi Road, Huaykwang, Bangkok 10310, Thailand Tel. +66 2716 6141-3 Fax +66 2716 6144 E-mail: [email protected] www.tci-thaijo.org/index.php/ThaiJSurg/index Royal College of Surgeons of Thailand Secretariat Office : Royal Golden Jubilee Building, 2 Soi Soonvijai, New Petchburi Road, Bangkok 10310, Thailand OFFICERS 2019 - 2021 President : Paisit Siriwittayakorn President-Elect : Pramook Mutirangura Vice President : Charnvate Satthaputh Secretary General : Sutdhachit Linananda Assistant Scretary : Pornprom Muangman Treasurer -

Operation Smile Charity Review Revised 2016
Charity Review Operation Smile Foundation (Thailand) Revised Date: 13/03/2017 Reviewers: Khun Terrence P. Weir,B Ec, CPA (Australia) Manu Pensawangwat, BA Charity Head: Executive Director: Ms. Christina Krause [email protected] Address: 12/2 Soi Methinivate, Sukumvit Soi 24 Klongtoey, Bangkok 10110 Telephone Numbers: +66 2-075-2700-2 Fax Number: +66 2 075-2703 Email Address: [email protected] Website http://www.operationsmile.or.th/ Charity Purpose: Operation Smile Thailand provides free surgeries to repair cleft lip, cleft palate and other facial deformities for children throughout the country. Operation Smile Thailand was registered as a Not-for-profit organization on Feb 11, 2002. Registration as a not-for-profit organization # Kor Tor 1112 Thailand Income Tax Exempt number # 636. Donations to Operation Smile Thailand are tax-deductible for Thai income tax payers. Approximately one in every 700 babies in Thailand is born with a cleft lip or cleft palate. The need for Operation Smile support for Cleft Lip and Cleft Palate surgeries in Thailand is well recognised as over 100 medical professionals in Thailand volunteer their time to perform surgical missions each year. Review Process: We visited the Operation Smile Thailand's office in Bangkok and reviewed the charity activities and discussion in activities details with Mr. Kevin J. Beauvais, Chairman and Ms. Christina Krause, Executive Director and K. Tatpisha Termthavorn, Resources Development Director. We gained a good understanding of the need the surgeries supported by Operation Smile Thailand and can see the high level of professionalism of the staff and transparency of the organisation. Background to the need: Operation Smile is an international non-profit medical charity founded in the U.S. -

129540588.Pdf
CoNTeNTS FOREWORD 03 A MESSAGE FROM OUR CO-DIRECTORS 04 HIV-NAT CLINICAL TRIALS NETWORK 05 INTERNATIONAL ADVISORY BOARD 2012 06 COLLABORATORS 07 OVERVIEW OF HIV-NAT STUDIES 10 PHARMACOKINETIC STUDIES 14 CLINICAL RESEARCH PROGRAMS IN CO-VIRAL & VIRAL INFECTIONS 15 CLINICAL RESEARCH PROGRAMS IN CO-TUBERCULOSIS INFECTIONS 19 LONG TERM COHORT ANALYSIS 20 STRATEGY STUDIES 23 NEW DRUG DEVELOPMENT 26 SEXUAL HEALTH 28 OTHERS 29 PEDIATRIC AND YOUTH RESEARCH PROGRAMS 31 HIV-NAT TEAMS 36 PUBLICATIONS & WRITINGS 40 PRESENTATIONS AT CONFERENCES AND MEETINGS 45 HIV-NAT RESEARCH LABORATORY 50 THE VACCINE & CELLULAR IMMUNOLOLGY LABORATORY 54 EDUCATION & DEVELOPMENT PROGRAM 56 15TH THE BANGKOK INTERNATIONAL SYMPOSIUM ON HIV MEDICINE 60 PARTNERSHIPS 62 COMMUNITY OUTREACH PROGRAMS 77 ACKNOWLEDGEMENTS 83 SPONSORS 84 2 H I V - N A T ANNUAL REPORT 2 0 1 2 Foreword It gives me great pride to present the 16th annual report rom the HIV Netherlands Australia Thailand Research Collaboration. Its pages highlight the dedication and proessionalism HIV-NAT Phan Wannamethee possesses in fghting HIV/AIDS through dierent Secretary General Tai Red Cross Society avenues o research, knowledge dissemination, activities and campaigns. HIV-NA started the year with the 15th Bangkok International Symposium on HIV Medicine in January, which was attended by more than 700 physicians rom all over the world. Additionally, various trainings on HIV/AIDS were organized throughout the year to disseminate knowledge and inormation on clinical research and HIV inection management to health care proessionals rom Tailand and 9 countries in the region. Troughout 2012 HIV-NA has stayed committed to improving HIV care in Tailand and Southeast Asia through cutting edge research and quality training or health care providers. -

Conservation of Heritage Healthcare Architecture; a Case Study at Siriraj Hospital, Bangkok Thailand
CONSERVATION OF HERITAGE HEALTHCARE ARCHITECTURE; A CASE STUDY AT SIRIRAJ HOSPITAL, BANGKOK THAILAND By Nantawat Sitdhiraksa A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY Program of Architectural Heritage Management and Tourism (International Program) Graduate School SILPAKORN UNIVERSITY 2011 CONSERVATION OF HERITAGE HEALTHCARE ARCHITECTURE; A CASE STUDY AT SIRIRAJ HOSPITAL, BANGKOK THAILAND By Nantawat Sitdhiraksa A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY Program of Architectural Heritage Management and Tourism (International Program) Graduate School SILPAKORN UNIVERSITY 2011 The Graduate School, Silpakorn University has approved and accredited the Thesis title of “Conservation of Heritage Healthcare Architecture; A Case Study at Siriraj Hospital, Bangkok Thailand” submitted by Mr.Nantawat Sitdhiraksa as a partial fulfillment of the requirements for the degree of Doctor of Philosophy in Architectural Heritage Management and Tourism ……........................................................................ (Assistant Professor Panjai Tantatsanawong,Ph.D.) Dean of Graduate School ............./..................../.......... The Thesis Advisor Dr. Donald Ellsmore The Thesis Examination Committee .................................................... Chairman (Professor Dr. Trungjai Buranasomphob) ............/......................../.............. .................................................... Member (Assistant Professor Pibul