Straightforward Conversion of Decoquinate Into Inexpensive Tractable New Derivatives with Significant Antimalarial Activities
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
2002 FSIS National Residue Program, Section 4
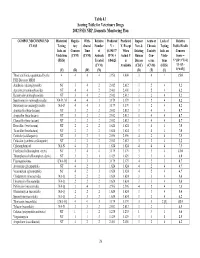
Table 4.1 Scoring Table for Veterinary Drugs 2002 FSIS NRP, Domestic Monitoring Plan COMPOUND/COMPOUND Historical Regula- With- Relative Predicted Predicted Impact Acute or Lack of Relative CLASS Testing tory drawal Number V = V, Except New & Chronic Testing Public Health Info. on Concern Time of (0.19437* When Existing Toxicity Info. on Concern Violations (CVM) (CVM) Animals R*N) + Actual V Human Con- Viola- Score = (FSIS) Treated 0.84625 is Disease cerns tions V*[(D+3*T)/4] (CVM) Available (CDC) (CVM) (FSIS) *{1+[(L- (V) (R) (W) (N) (D) (T) (L) 1)*0.05]} Those antibiotics quantitated by the 4 4 4 4 3.956 4.000 3 4 1 15.0 FSIS Bioassay MRM Amikacin (aminoglycoside) NT 3 4 2 2.012 2.012 3 2 4 5.2 Apramycin (aminoglycoside) NT 4 4 2 2.401 2.401 3 2 4 6.2 Kanamycin (aminoglycoside) NT 3 4 2 2.012 2.012 3 2 4 5.2 Spectinomycin (aminoglycoside) NA-D, M 4 4 3 3.179 3.179 3 2 4 8.2 Streptomycin (aminoglycoside) NA-D 4 4 3 3.179 3.179 3 2 4 8.2 Amoxicillin (beta-lactam) NT 3 2 2 2.012 2.012 3 4 4 8.7 Ampicillin (beta-lactam) NT 3 2 2 2.012 2.012 3 4 4 8.7 Cloxacillin (beta-lactam) NT 3 2 2 2.012 2.012 3 4 4 8.7 Hetacillin (beta-lactam) NT 2 2 2 1.624 1.624 3 4 4 7.0 Ticarcillin (beta-lactam) NT 2 2 2 1.624 1.624 3 4 4 7.0 Ceftiofur (cefalosporin) NT 3 2 3 2.596 2.596 4 2 4 7.5 Cefazolin (synthetic cefalosporin) NT 3 2 2 2.012 2.012 3 2 4 5.2 Chloramphenicol NA-N 4 2 1 1.624 1.624 4 4 4 7.5 Florfenicol (chloramphen. -

2019-6654 Final Report of an Audit Carried out In
Ref. Ares(2019)7826731 - 19/12/2019 EUROPEAN COMMISSION DIRECTORATE-GENERAL FOR HEALTH AND FOOD SAFETY Health and food audits and analysis DG(SANTE) 2019-6654 FINAL REPORT OF AN AUDIT CARRIED OUT IN BELARUS FROM 13 TO 24 MAY 2019 IN ORDER TO EVALUATE THE CONTROL OF RESIDUES AND CONTAMINANTS IN LIVE ANIMALS AND ANIMAL PRODUCTS INCLUDING CONTROLS ON VETERINARY MEDICINAL PRODUCTS Executive Summary This report describes the outcome of an audit carried out in Belarus from 13 to 24 May 2019 as part of the European Commission’s Directorate-General for Health and Food Safety planned work programme. The objective of the audit was to evaluate the effectiveness of official controls on residues and contaminants in live animals and animal products eligible for export to the European Union (EU). The audit assessed the implementation of the residue monitoring plan and also covered the authorisation, distribution and use of veterinary medicinal products, given that these areas have an impact on the monitoring of residues. Attention was also paid to examining the implementation of corrective actions indicated in response to specific recommendations made in the report of the previous residues audit to Belarus. The planning of the residue monitoring largely follows the principles of Directive 96/23/EC and covers for the most part an appropriate range of substances. The plan is nevertheless weakened by the fact that action levels for several substances across all commodities (including those for which listing has been requested) are not aligned with EU maximum residue limits, thus the plan would not be sufficient to demonstrate that commodities eligible for export to the EU would comply with such limits where they are lower that national limits. -

The Organic Chemistry of Drug Synthesis
The Organic Chemistry of Drug Synthesis VOLUME 2 DANIEL LEDNICER Mead Johnson and Company Evansville, Indiana LESTER A. MITSCHER The University of Kansas School of Pharmacy Department of Medicinal Chemistry Lawrence, Kansas A WILEY-INTERSCIENCE PUBLICATION JOHN WILEY AND SONS, New York • Chichester • Brisbane • Toronto Copyright © 1980 by John Wiley & Sons, Inc. All rights reserved. Published simultaneously in Canada. Reproduction or translation of any part of this work beyond that permitted by Sections 107 or 108 of the 1976 United States Copyright Act without the permission of the copyright owner is unlawful. Requests for permission or further information should be addressed to the Permissions Department, John Wiley & Sons, Inc. Library of Congress Cataloging in Publication Data: Lednicer, Daniel, 1929- The organic chemistry of drug synthesis. "A Wiley-lnterscience publication." 1. Chemistry, Medical and pharmaceutical. 2. Drugs. 3. Chemistry, Organic. I. Mitscher, Lester A., joint author. II. Title. RS421 .L423 615M 91 76-28387 ISBN 0-471-04392-3 Printed in the United States of America 10 987654321 It is our pleasure again to dedicate a book to our helpmeets: Beryle and Betty. "Has it ever occurred to you that medicinal chemists are just like compulsive gamblers: the next compound will be the real winner." R. L. Clark at the 16th National Medicinal Chemistry Symposium, June, 1978. vii Preface The reception accorded "Organic Chemistry of Drug Synthesis11 seems to us to indicate widespread interest in the organic chemistry involved in the search for new pharmaceutical agents. We are only too aware of the fact that the book deals with a limited segment of the field; the earlier volume cannot be considered either comprehensive or completely up to date. -

United States Patent (19) 11) Patent Number: 5,001,115 Sloan (45) Date of Patent: Mar
United States Patent (19) 11) Patent Number: 5,001,115 Sloan (45) Date of Patent: Mar. 19, 1991 54 PRODRUGS OF BIOLOGICALLY ACTIVE Primary Examiner-Mukund J. Shah HYDROXYAROMATIC COMPOUNDS Assistant Examiner-E. L. Ward Attorney, Agent, or Firm-Kerkam, Stowell, Kondracki (75 Inventor: Kenneth B. Sloan, Gainesville, Fla. & Clarke 73 Assignee: University of Florida, Gainesville, Fla, (57) ABSTRACT (21) Appl. No.: 352,919 Prodrugs of bio-active hydroxyaromatic drugs having 22 Filed: May 17, 1989 the structural formula: A pharmaceutically acceptable prodrug of a biologi (51] Int. Cl. ............... A61K 31/70; A61K 31/595; cally active, therapeutically effective hydroxyaromatic A61K 31/535; A61K 31/255 52 U.S. C. ..................................... 514/34: 514/289; drug, said prodrug being selected from the group con 514/169; 514/373; 514/222.8; 514/328; sisting of, (A) compounds having the structural for 514/360; 514/603; 514/417; 514/425; 514/518; mula: 546/44; 54.6/176; 546/75; 544/2; 536/64; 548/123; 548/209; 548/256; 548/417 DRUG-O-CR'R''-2) 58 Field of Search ............... 514/169,289, 373, 417, 514/425, 222.8, 328,360, 518, 603, 34; 536/64; wherein: 564/82, 155; 552/626; 546/44, 176, 75; 544/2; DRUG -O- is the hydroxyaromatic O-dehydro 548/123, 209, 256, 477, 595 residue of said drug; 56 References Cited R" and R' may be the same or different and may be H, PUBLICATIONS alkyl, aryl or electron withdrawing groups; 2 is a displaceable leaving group; and Katritzky, et al. J. Chem. Soc. -

Swedres-Svarm 2004
SVARM2004 Swedish Veterinary Antimicrobial Resistance Monitoring Preface .............................................................................................4 Summary ..........................................................................................6 Sammanfattning ...............................................................................7 Use of antimicrobials .........................................................................8 Resistance in zoonotic bacteria ......................................................12 Salmonella ..........................................................................................................12 Campylobacter ...................................................................................................15 Resistance in indicator bacteria ......................................................17 Escherichia coli ...................................................................................................17 Enterococcus .....................................................................................................20 Resistance in animal pathogens ......................................................28 Pig ......................................................................................................................28 Cattle ..................................................................................................................29 Horse ..................................................................................................................30 -

Danmap 2006.Pmd
DANMAP 2006 DANMAP 2006 DANMAP 2006 - Use of antimicrobial agents and occurrence of antimicrobial resistance in bacteria from food animals, foods and humans in Denmark Statens Serum Institut Danish Veterinary and Food Administration Danish Medicines Agency National Veterinary Institute, Technical University of Denmark National Food Institute, Technical University of Denmark Editors: Hanne-Dorthe Emborg Danish Zoonosis Centre National Food Institute, Technical University of Denmark Mørkhøj Bygade 19 Contents DK - 2860 Søborg Anette M. Hammerum National Center for Antimicrobials and Contributors to the 2006 Infection Control DANMAP Report 4 Statens Serum Institut Artillerivej 5 DK - 2300 Copenhagen Introduction 6 DANMAP board: National Food Institute, Acknowledgements 6 Technical University of Denmark: Ole E. Heuer Frank Aarestrup List of abbreviations 7 National Veterinary Institute, Tecnical University of Denmark: Sammendrag 9 Flemming Bager Danish Veterinary and Food Administration: Summary 12 Justin C. Ajufo Annette Cleveland Nielsen Statens Serum Institut: Demographic data 15 Dominique L. Monnet Niels Frimodt-Møller Anette M. Hammerum Antimicrobial consumption 17 Danish Medicines Agency: Consumption in animals 17 Jan Poulsen Consumption in humans 24 Layout: Susanne Carlsson Danish Zoonosis Centre Resistance in zoonotic bacteria 33 Printing: Schultz Grafisk A/S DANMAP 2006 - September 2007 Salmonella 33 ISSN 1600-2032 Campylobacter 43 Text and tables may be cited and reprinted only with reference to this report. Resistance in indicator bacteria 47 Reprints can be ordered from: Enterococci 47 National Food Institute Escherichia coli 58 Danish Zoonosis Centre Tecnical University of Denmark Mørkhøj Bygade 19 DK - 2860 Søborg Resistance in bacteria from Phone: +45 7234 - 7084 diagnostic submissions 65 Fax: +45 7234 - 7028 E. -

U.S. National Residue Program: 2015 Residue Sampling Plans (Blue Book)
UNITED STATES National Residue Program for Meat, Poultry, and Egg Products 2015 Residue Sampling Plans United States Department of Agriculture Food Safety and Inspection Service Office of Public Health Science March 2015 Table of Contents Table of Contents ............................................................................................................................. ii Preface ............................................................................................................................................ iii Contacts and Comments ................................................................................................................. iii Acknowledgements ......................................................................................................................... iii Principal Authors ............................................................................................................................ iii Acronyms ........................................................................................................................................ iv Introduction ...................................................................................................................................... 1 Overview of the Sampling Plans ...................................................................................................... 5 Domestic Sampling Plan ..................................................................................................... 5 Import Reinspection Sampling -

Wo 2010/059253 A2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 27 May 2010 (27.05.2010) WO 2010/059253 A2 (51) International Patent Classification: 02140 (US). BERSHTEYN, Anna [US/US]; 25 Linden A61K 9/16 (2006.01) A61K 31/711 (2006.01) Avenue, Apartment 6, Somerville, MA 02143 (US). A61K 9/127 (2006.01) A61P 35/00 (2006.01) A61K 31/7105 (2006.01) (74) Agent: TREVISAN, Maria, A.; Wolf, Greenfield & Sacks, P.C , Federal Reserve Plaza, 600 Atlantic Avenue, (21) International Application Number: Boston, MA 02210-2206 (US). PCT/US2009/006290 (81) Designated States (unless otherwise indicated, for every (22) International Filing Date: kind of national protection available): AE, AG, AL, AM, 24 November 2009 (24.1 1.2009) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, (25) Filing Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (26) Publication Language: English HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (30) Priority Data: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 61/200,1 60 24 November 2008 (24.1 1.2008) US NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, (71) Applicant (for all designated States except US): MAS- SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, SACHUSETS INSTITUTE OF TECHNOLOGY [US/ TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -
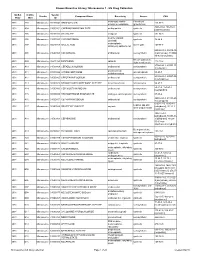
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

Arasites of Cattle
arasites of Cattle CONTENTS 1 Stages in the gut and faeces . ............ 24 • 2 Stages in the blood and circulatory system . .................... 55 • 3 Stages in the urogenital system ........ 83 . 4 Stages in internaiorgans . ............... 85 4.1 Locomotory system .................. 85 4.7 .7 Muscles ...................... 85 4.7.2 Tendons . .................... 90 4.2 Liver ............................. 90 4.3 Respiratory system ................... 97 4.4 Abdominal cavity .................. 101 4.5 Pancreas ......................... 102 4.6 Central nervous system .............. 103 • 5 Stages on the body surface . ............ 105 5.1 Skin and co at ..................... 105 5.2 Eyes ............................. 143 J. Kaufmann, Parasitic Infections of Domestic Animals © Springer Basel AG 1996 1 Stages In the gut and taeces , Stages in the gut and faeces and para lysis. Death can occur rapidly, mainly in calves. Another form of coccidio sis is characterized by persisting, non-ha em orrhagic diarrhoea with continuous weight PROTOZOA loss until cachexia. This condition may last • Protozoa oocysts found in the faeces . .. 24 for several weeks. Animals that survive severe illness can have significant weight HELMINTHS loss that is not quickly regained, or can • Trematoda eggs found in the remain permanently stunted. faeces and adult trematodes living in the gastrointestinal tract . ..... .. 29 Significance: E. hovis and E. zuerni are most commonly involved in c1inical coccidiosis • Cestoda eggs found in the faeces and adult cestodes living in the of cattle. gastrointestinal tract ...... .. ... 32 Diagnosis: Clinical signs and extremely high • Nematoda eggs found in the faeces, numbers of oocysts per gram of faeces adult nematodes living in the gastro (50,000-500,000). intestinal tract and first-stage Therapy: The drugs that are commonly used larvae of Dictyocaulus viviparus . -

American Canine Hepatozoonosis
CONTINUING EDUCATION FOR VETERINARIANS ® CE Article www.CompendiumVet.com American Canine Hepatozoonosis Emily M. Medici, DVM Oklahoma State University Johanna Heseltine, DVM, MS, DACVIM Canada West Veterinary Specialists and Critical Care Hospital ABSTRACT: American canine hepatozoonosis (ACH) is a debilitating disease caused by Hepatozoon americanum and transmitted by ingestion of oocyst-containing Amblyomma maculatum ticks . A history of incomplete response to common antibiotics , together with physical findings of fever and muscle wasting and laboratory findings of leukocytosis and periosteal proliferation , should prompt testing for ACH. Muscle biopsy and whole-blood polymerase chain reaction are currently the most reliable methods of diagnosis. Treatment consists of a 2-week course of trimethoprim –sulfonamide, clindamycin, and pyrimethamine , followed by a 2-year course of decoquinate. Relapses are common, but prognosis is fair overall. he cause of American canine hepato - GEOGRAPHIC DISTRIBUTION zoonosis (ACH) is Hepatozoon ameri - AND EPIDEMIOLOGY T canum, a protozoan transmitted by ACH has been reported in Texas, Louisiana, ingestion of Amblyomma maculatum ticks , or Alabama, Mississippi, Oklahoma, Georgia, Gulf Coast ticks , that contain the oocysts of the Tennessee , and Florida. 3,5,8–10 The geographic organism (Figure 1). Canine hepatozoonosis distribution of H. americanum is directly related was first recognized in North America in 1978 to the distribution of the Gulf Coast tick, in Texas .1 All infections in North America were which is the obligate definitive host and the originally thought to be caused by a more viru - source of infective oocysts of the protozoan. lent strain of Hepatozoon canis, the “Texas This tick , which was once confined to the Texas strain.” 2–5 In 1997, however, based on clinical, Gulf Coast , has now expanded its range as far serologic , and pathologic features ; gamont mor - north as southern Kansas and Kentucky. -

Antimicrobial Feed Additives
SOU 1997:132 141 5 Toxicological and related effects of antibacterial feed additives 5.1 Introduction As animals are fed AFA for long periods of time, knowledge on bioaccumulation, chronic toxicity and problems associated with residues is imperative. AFA are defined chemical substances and might as such have toxic or allergenic properties. According to directive 70/524/EEC and the guidelines provided in 94/40/EC, satisfactory information relating to these topics has to be presented before approval. Toxic effects, if any, could be seen either in target animal species (or non- target animals as a result of accidental intake) or in humans. Animals and humans may be exposed via residues in animal products and humans also when handling products containing the substances. If the product has unwanted properties, such as organ toxicity, mutagenicity or allergenicity, both ways of exposure could be harmful. Some AFA are poorly absorbed from the gut. Residues are therefore normally not an issue for these substances. Other AFA such as ardacin and avilamycin are absorbed to some extent, and tylosin, spiramycin, olaquindox and carbadox are well absorbed after oral administration (FAO/WHO, 1991; MagnussenETAL, 1991; FAO/WHO, 1994). Further, the possibility of accumulation in the animal must also be addressed. In the following, some information with relevance to the topic will be discussed. Toxicological aspects of coccidiostats, nitroimidazoles and ionophoric AFA are discussed in chapter 7. 5.2 Toxicity for target species With a few exceptions, AFA are not expected to cause toxic reactions in target species at the levels permitted. Bacitracin is nephrotoxic (Prescott and Baggot, 1993), but is not absorbed from the gut.