Lysosomal Storage Disorders Screen Interpretive Algorithm
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neuroradiologic Findings in Fucosidosis, a Rare Lysosomal Storage Disease
Neuroradiologic Findings in Fucosidosis, a Rare Lysosomal Storage Disease James M. Provenzale, Daniel P. Barboriak, and Katherine Sims Summary: Fucosidosis is a rare lysosomal storage disorder with vacuolated secondary lysosomes containing some fine the clinical features of mental retardation, cardiomegaly, dysos- fibrillar material and lamellated membrane structures. tosis multiplex, progressive neurologic deterioration, and early A magnetic resonance (MR) examination at the age of death. The neuroradiologic findings in two patients are reported, 10 years showed confluent regions of hyperintense signal and include abnormalities within the globus pallidus (both pa- on T2-weighted images in the periventricular regions, tients) and periventricular white matter (one patient). most prominent surrounding the atria of the lateral ventri- cles. Hyperintense signal was noted in the globus pallidus Index terms: Brain, metabolism; Degenerative disease, Pediatric on T1-weighted images, with corresponding hypointense neuroradiology signal on T2-weighted images (Fig 1). Fucosidosis is a rare metabolic disorder caused by decreased amounts of the enzyme Case 2 a-L-fucosidase, which results in the accumula- A 2-year-old boy was examined for speech delay and tion of fucose-rich storage products within psychomotor retardation. He was born at term after a many organs, including the brain. Patients with normal pregnancy, and appropriate development oc- this disorder usually have delayed intellectual curred during the first year of life. However, by age 2 years, and motor development, and often die within the patient had not developed speech, exhibited autistic the first decade of life. Computed tomographic behavior, and performed motor tasks poorly. Physical ex- (CT) findings have been reported in a few cases amination findings included coarsened facial features, nar- (1, 2). -

File Download
Radiological and clinical characterization of the lysosomal storage disorders: non-lipid disorders Minzhi Xing, Emory University E I Parker, Emory University A Moreno-De-Luca, Geisinger Health System Elie Harmouche, Emory University Michael Terk, Emory University Journal Title: British Journal of Radiology Volume: Volume 87, Number 1033 Publisher: British Institute of Radiology | 2014-01-01, Pages 20130467-20130467 Type of Work: Article | Final Publisher PDF Publisher DOI: 10.1259/bjr.20130467 Permanent URL: https://pid.emory.edu/ark:/25593/pr2p5 Final published version: http://dx.doi.org/10.1259/bjr.20130467 Copyright information: © 2014 The Authors. This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/), which permits distribution of derivative works, making multiple copies, distribution, public display, and publicly performance, provided the original work is properly cited. This license requires credit be given to copyright holder and/or author, copyright and license notices be kept intact. Accessed September 24, 2021 3:09 PM EDT BJR © 2014 The Authors. Published by the British Institute of Radiology Received: Revised: Accepted: doi: 10.1259/bjr.20130467 28 July 2013 29 October 2013 8 November 2013 Cite this article as: Xing M, Parker EI, Moreno-De-Luca A, Harmouche E, Terk MR. Radiological and clinical characterization of the lysosomal storage disorders: non-lipid disorders. Br J Radiol 2014;87:20130467. REVIEW ARTICLE Radiological -

Fast Urinary Screening of Oligosaccharidoses by MALDI-TOF/TOF Mass Spectrometry
Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry. Laurent Bonesso, Monique Piraud, Céline Caruba, Emmanuel van Obberghen, Raymond Mengual, Charlotte Hinault To cite this version: Laurent Bonesso, Monique Piraud, Céline Caruba, Emmanuel van Obberghen, Raymond Mengual, et al.. Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry.. Orphanet Journal of Rare Diseases, BioMed Central, 2014, 9 (1), pp.19. 10.1186/1750-1172-9-19. inserm- 00945684 HAL Id: inserm-00945684 https://www.hal.inserm.fr/inserm-00945684 Submitted on 12 Feb 2014 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Bonesso et al. Orphanet Journal of Rare Diseases 2014, 9:19 http://www.ojrd.com/content/9/1/19 RESEARCH Open Access Fast urinary screening of oligosaccharidoses by MALDI-TOF/TOF mass spectrometry Laurent Bonesso1, Monique Piraud5, Céline Caruba1, Emmanuel Van Obberghen1,2,3,4, Raymond Mengual1† and Charlotte Hinault1,2,3,4*† Abstract Background: Oligosaccharidoses, which belong to the lysosomal storage diseases, are inherited metabolic disorders due to the absence or the loss of function of one of the enzymes involved in the catabolic pathway of glycoproteins and indirectly of glycosphingolipids. -

Fucosidosis: Ultrastructural Study of Conjunctiva and Skin and Enzyme Analysis of Tears
Fucosidosis: ultrastructural study of conjunctiva and skin and enzyme analysis of tears /. Libert,9 F. Van Hoof," and M. Tondeur*** Conjunctival and skin biopsies from two new patients with fucosidosis were studied by electron microscopy. In both tissues, the connective tissue cells and the capillary endothelial cells were filled with single membrane limited inclusions of two types: (1) Clear inclusions containing a fibrillogranular reticulum. (2) Dark inclusions with a dense granular material. Specific stainings in ultrastructure suggest that these inclusions contain oligosaccharide chains. The ultrastructural aspect is characteristic for fucosidosis. Enzyme studies on tears realize an easy and secure technique for the diagnosis of the disease. -Lhe interest of conjunctival biopsy in the io-i5 Most of the lysosomal acid hydrolases study of storage diseases is now abun- can be assayed in tears, and their defi- dantly documented. Pathologic changes ciency allows the diagnosis of several other have been demonstrated in the systemic inborn lysosomal diseases.16"18 mucopolysaccharidoses,1 several mucolip- Fucosidosis, an autosomal lysosomal dis- idoses,2"5 Fabry's disease,6'7 Niemann- ease, is characterized by the absence or the Pick's disease,8 and cystinosis.9 profound deficiency of a-L-fucosidase,19"21 The usefulness of tears for the enzymatic which results in the widespread accumula- screening of inborn lysosomal diseases was tion of fucose-containing glycosphingolip- recognized about two years ago and ap- ids, oligosaccharides, and polysaccharides, plied to Tay-Sachs and Fabry's diseases.7' together with an increased excretion of fucosides in urine.22 Clinically, a severe and a mild phenotype have been distin- From the Departments of Ophthalmology" and guished.23' 24 The patients suffering from Pediatrics,00" Hopital Saint-Pierre, Universite Libre de Bruxelles, and from the International the severe form present a progressive psy- Institute of Cellular and Molecular Patholo- chomotor retardation and a moderate gy,00 Universite Catholique de Louvain. -

Lysosomal Diseases
THE GLYCOPROTEINOSES: SECOND INTERNATIONAL WORKSHOP ON ADVANCES IN PATHOGENESIS AND THERAPY JULY 26-27, 2007 ANN ARBOR, MICHIGAN Scientific Conference Day 1 Morning Sessions, July 26 7:30-8:15 Continental Breakfast 8:15-8:30 Welcome and Introductions, Opening Remarks – Goals of the conference, progress since the last conference - Steven Walkley (with additional comments by officials from ISMRD, NINDS, ORD) 8:30-9:15 Plenary Address Cross-Correction: Looking Back, Looking Forward – Elizabeth Neufeld Session 1: Animal Models, Pathogenesis, Experimental Therapies (Chair, Mark Haskins) α-Mannosidosis 9:15-9:35 Enzyme replacement therapy for α-Mannosidosis - Judith Blanz 9:35-9:55 α-Mannosidosis in the Guinea Pig – Pathophysiological, behavioral, and treatment issues - John Hopwood 10:00-10:15 Break α-Mannosidosis (continued) 10:15-10:35 Strategies for translating gene therapy in the brain from rodents to clinical use - John Wolfe 10:35-10:55 Quantitative Imaging of gray matter disease associated with feline α- mannosidosis – Charles Vite β-Mannosidosis 10:55-11:15 β-Mannosidosis mice – A model for the human disease? – Karen Friderici Fucosidosis 11:15-11:30 Intrathecal enzyme infusion therapy in canine fucosidosis – Gauthami Kondagari 11:30-12:00 General Discussion 12:00-1:30 Lunch Afternoon Session, July 26 1:30-2:15 Special Lecture: The Glycoproteinoses: Limited Progress in Molecular Insight still Surpasses Present-day Treatment Efficacy – Jules Leroy Session 2: Animal Models, Pathogenesis, Experimental Therapies (Chair, John Wolfe) -

4Th Glycoproteinoses International Conference Advances in Pathogenesis and Therapy
Program & Abstracts 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ADVANCES IN PATHOGENESIS AND THERAPY ISMRD ST. LOUIS, MISSOURI, UNITED STATES Program & Abstracts I SM R D ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD would like to say A Very Special Thank You to the following organizations and companies who have very generously given donations and sponsorship to support the 4th International Conference on Glycoproteinoses THE PRENILLE EDWARD MALLINCKRODT FOUNDATION JR FOUNDATION MARK HASKINS I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD is very proud to display 10 featured Expression of Hope artworks to be Auctioned at the Gala Dinner. These beautiful prints are from Genzyme’s featured Artwork selection. Contents Welcome 1 SCIENTIFIC COMMITTEE: Stuart Kornfeld ISMRD Mission & Governance 3 (Chair, Scientifi c Planning Committee) Steve Walkley Sara Cathey ISMRD General Information 5 Richard Steet Sean Thomas Ackley, Philippines Miriam Storchli, Switzerland Alessandra d’Azzo ‘Hope’ by Sarah Noble, New Zealand Scientifi c Program 9 FAMILY CONFERENCE COMMITTEE: Family Program for Mucolipidosis 11 Jenny Noble (Conference Organiser) Jackie James (Conference Organiser Family Program For Alpha Mannosidosis /Sialidosis/ 13 - St. Louis) Fucosidosis/Aspartylglucosaminuria Mark Stark John Forman ‘All around the world’ by Zih Yun Li , Taiwan Childrens Program 16 Susan Kester Carolyn Paisley-Dew Tish Adkins Abstracts 17 Sara DeAngelis, Russia Gayle Rose, United States Speaker Profi les 60 Delegates 81 Helen Walker, Australia Nicklas Harkins, Canada Naomi Arai, Japan David Wentworth, Serbia I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts On behalf of the Scientifi c Planning Committee, I want to extend a warm welcome to all the investigators and Welcome! families who have traveled to St. -

Mucopolysaccharidoses and Mucolipidoses
J Clin Pathol: first published as 10.1136/jcp.s3-8.1.64 on 1 January 1974. Downloaded from J. clin. Path., 27, Suppl. (Roy. Coll. Path.), 8, 64-93 Mucopolysaccharidoses and mucolipidoses F. VAN HOOF From the Laboratoire de Chimie Physiologique, Universite Catholique de Louvain, and International Institute of Cellular and Molecular Pathology, Brussels, Belgium Many different syndromes are classified as muco- THE CHEMICAL ERA polysaccharidoses, and, despite remarkable progress Chemical studies, performed mainly by groups in the biochemical understanding of these diseases, working with A. Dorfman, in Chicago and K. much remains to be learned and many cases still Meyer, in New York, have provided most of the escape classification. new knowledge in the field by analysis of tissue and Mucopolysaccharidoses are inborn storage dis- urinary mucopolysaccharides in patients (Dorfman eases, characterized by a complex accumulation of and Lorincz, 1957; Meyer, Grumbach, Linker, and mucopolysaccharides and of glycolipids within the Hoffman, 1958; Meyer, Hoffman, Linker, lysosomes. Sixteen human diseases correspond to Grumbach, and Sampson, 1959). These provided the this definition, of which nine have been presently basis for the subdivision of the 'Hurler syndrome' explained by the deficiency of an acid hydrolase. into six subgroups (McKusick, Kaplan, Wise, They are somewhat arbitrarily divided into muco- Hanley, Suddarth, Sevick, and Maumanee, 1965). polysaccharidoses and mucolipidoses. In muco- The possibility that mucopolysaccharidoses could polysaccharidoses, mucopolysaccharides are the result from an excessive biosynthesis of muco- main storage substances, although an abnormal polysaccharides was suggested by Matalon and accumulation of complex lipids is practically always Dorfman (1966). copyright. disclosed at least by the ultiastructural examination. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

Alpha-Fucosidosis ¬タモ Two Brothers Presenting with Dysostosis Multiplex
The Egyptian Journal of Medical Human Genetics (2016) 17, 243–246 HOSTED BY Ain Shams University The Egyptian Journal of Medical Human Genetics www.ejmhg.eg.net www.sciencedirect.com CASE REPORT Alpha-fucosidosis – Two brothers presenting with dysostosis multiplex Rimshah Shaukat a, Syed Musa Raza a, Zabedah Md. Yuns b, Affandi Omar b, Bushra Afroze c,* a Aga Khan Medical University, Karachi, Pakistan b Institute of Medical Research, Kuala Lumpur, Malaysia c Department of Paediatrics & Child Health, Aga Khan University Hospital, Karachi, Pakistan Received 5 November 2015; accepted 30 November 2015 Available online 30 December 2015 KEYWORDS Abstract a-Fucosidosis is a rare inherited neuro-degenerative disorder causing progressive neuro- a Alpha-fucosidosis; logical deterioration leading to early death. Definitive diagnosis requires -fucosidase enzyme assay Pakistani patients; or FUCA1 gene testing, which being expensive limits the definitive diagnosis in resource limited Dysostosis multiplex countries. We present two siblings with classic symptoms, radiological and MRI brain findings sug- gestive of a-fucosidosis and a clinical approach to reach to the diagnosis. Ó 2015 The Authors. Production and hosting by Elsevier B.V. on behalf of Ain Shams University. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/). 1. Introduction a-fucosidosis from Pakistan, focusing on radiological findings as a diagnostic tool to develop an approach to a patient with a-Fucosidosis (OMIM # 230000) is an autosomal recessive dysostosis multiplex, in neuro-degenerative disorders including a lysosomal storage disorder caused by the deficiency of fucosi- -fucosidosis (Fig. 1). -

A Case of Type I Sialidosis with Osteonecrosis Revealing a New
Original Article Journal of Inborn Errors of Metabolism &Screening 1–3 A Case of Type I Sialidosis With ª The Author(s) 2014 Reprints and permission: Osteonecrosis Revealing a New sagepub.com/journalsPermissions.nav DOI: 10.1177/2326409814543468 Mutation in NEU1 iem.sagepub.com Geoffrey Urbanski1,2, Soumeya Bekri3, Magalie Barth2,4, Christophe Verny4, and Christian Lavigne1,2 Abstract Sialidosis is a rare lysosomal storage disease. The 2 forms described are as follows: the early-onset form, or type II, presents with dysostosis multiplex, while the late-onset form, or type I, does not involve bone in the literature. We report the case of a 42-year- old woman with type I sialidosis who presents with osteonecrosis of both humeral and femoral heads. Molecular study reveals a never listed mutation of NEU1 in exon 5, p.Gly273Asp (c.818G>A), and a second known missense mutation. Keywords sialidosis, bone involvement, NEU1 Introduction developed, at the age of 18 years, a rapidly progressive severe bilateral visual defect leading to blindness. At that time, the Sialidosis (Online Mendelian Inheritance in Man [OMIM] ophthalmologic examination revealed bilateral cherry red spot 256550) is a rare lysosomal storage disease,1 with an estimated in the macula, evolving to macular and optic atrophy associated incidence of 1 in 4 200 000 live births, and it belongs to the group with bilateral cataract. At the age of 32 years, she developed of oligosaccharidoses. Sialidosis is caused by to the recessively myoclonus and epilepsy as grand mal seizure. Myoclonus inherited deficiency of N-acetyl-a-neuraminidase, an acid affected all 4 limbs, but prevailed in the upper limbs, and hydrolase expressed from the gene NEU1, which is located in increased with menstrual cycle and anxiety. -
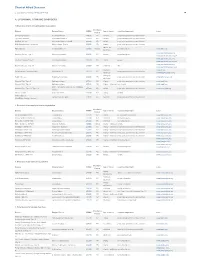
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none -

The Lysosomal Sialic Acid Transporter Sialin Is Required for Normal CNS Myelination
The Journal of Neuroscience, December 9, 2009 • 29(49):15355–15365 • 15355 Neurobiology of Disease The Lysosomal Sialic Acid Transporter Sialin Is Required for Normal CNS Myelination Laura M. Prolo,1 Hannes Vogel,2 and Richard J. Reimer1 1Department of Neurology and Neurological Sciences and Graduate Program in Neuroscience and 2Departments of Pathology and Pediatrics, Stanford University School of Medicine, Stanford, California 94305 Salla disease and infantile sialic acid storage disease are autosomal recessive lysosomal storage disorders caused by mutations in the gene encoding sialin, a membrane protein that transports free sialic acid out of the lysosome after it is cleaved from sialoglycoconjugates undergoing degradation. Accumulation of sialic acid in lysosomes defines these disorders, and the clinical phenotype is characterized by neurodevelopmental defects, including severe CNS hypomyelination. In this study, we used a sialin-deficient mouse to address how loss of sialin leads to the defect in myelination. Behavioral analysis of the sialin ؊/؊ mouse demonstrates poor coordination, seizures, and premature death. Analysis by histology, electron microscopy, and Western blotting reveals a decrease in myelination of the CNS but normal neuronal cytoarchitecture and normal myelination of the PNS. To investigate potential mechanisms underlying CNS hypomyeli- nation, we studied myelination and oligodendrocyte development in optic nerves. We found reduced numbers of myelinated axons in optic nerves from sialin ؊/؊ mice, but the myelin that was present appeared grossly normal. Migration and density of oligodendrocyte precursorcellswerenormal;however,amarkeddecreaseinthenumberofpostmitoticoligodendrocytesandanassociatedincreaseinthe number of apoptotic cells during the later stages of myelinogenesis were observed. These findings suggest that a defect in maturation of cells in the oligodendrocyte lineage leads to increased apoptosis and underlies the myelination defect associated with sialin loss.