Lysosomal Disease Network Consortiapedia.Fastercures.Org
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none -

Human Xc-N-Acetylgalactosaminidase Qx-NAGA)
4585 JMed Genet 1996;33:458-464 Human xc-N-acetylgalactosaminidase Qx-NAGA) deficiency: new mutations and the paradox J Med Genet: first published as 10.1136/jmg.33.6.458 on 1 June 1996. Downloaded from between genotype and phenotype J L M Keulemans, A J J Reuser, M A Kroos, R Willemsen, M M P Hermans, A M W van den Ouweland, J G N de Jong, R A Wevers, W 0 Renier, D Schindler, M J Coll, A Chabas, H Sakuraba, Y Suzuki, 0 P van Diggelen Abstract reported the second independent case of a- Up to now eight patients with a-NAGA NAGA deficiency with an entirely different deficiency have been described. This in- clinical phenotype. This patient had a late onset cludes the newly identified patient re- disease with slight facial coarseness, dis- ported here who died unexpectedly aged seminated angiokeratoma, and mild intellectual II years of hypoxia during convulsions; impairment (IQ= 70), but without neuro- necropsy was not performed. logical symptoms. Unlike the infantile cases, Three patients have been genotyped pre- this patient had prominent vacuolisation in all viously and here we report the mutations dermal cells, most prominently in vascular and in the other five patients, including two lymphatic endothelial cells and eccrine sweat new mutations (S160C and E193X). The gland cells, but also in dermal neural cells and newly identified patient is consanguineous fibroblasts.' The glomerular endothelial cells with the first patients reported with a- but not the epithelial kidney cells are involved NAGA deficiency and neuroaxonal dys- and also blood lymphocytes are vacuolised.6 trophy and they all had the a-NAGA geno- These three patients shared, however, the type E325KIE325K. -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

Lysosomal Storage Disorders: Urine Screening
2460 Mountain Industrial Boulevard | Tucker, Georgia 30084 Phone: 470-378-2200 or 855-831-7447 | Fax: 470-378-2250 eglgenetics.com Lysosomal Storage Disorders: Urine Screening Test Code: BLSDS Turnaround time: 2 weeks (GAGs performed Fri 10 am / Oligos performed Fri 3 pm) CPT Codes: 82542 x1, 82570 x1, 83864 x1, 84275 x1, 84375 x1, 84377 x1 Condition Description Notice: This test has been discontinued EGL no longer accepts samples for this test. For questions, please call: 470-378-2200 In glycoprotein storage diseases (GSDs), certain subtypes of congenital disorders of glycosylation (CDGs), and in the mucolipidoses, there is an accumulation of oligosaccharides, free glycans, glycoamino acids, glycolipid and glycopeptide in the urine. Glycoprotein storage diseases are genetic conditions caused by the body's inability to produce specific enzymes. Normally, the body uses enzymes to process, break down and recycle materials in cells. In individuals with GSD and related diseases, the missing or insufficient enzyme prevents the proper processing and recycling process. This results in the storage of materials, called oligosaccharides or free glycans and glycoamino acids in virtually every cell of the body. As a result, cells do not perform properly and may cause progressive damage throughout the body, including the heart, bones, joints, respiratory system, immune system and central nervous system. While the disease may or may not be apparent at birth, signs and symptoms develop with age as more cells become damaged by the accumulation of materials. The symptoms of these diseases may vary based on syndrome type, and in some cases may resemble a mucopolysaccharidosis. This urinary oligosaccharide and glycan screening uses mass spectrometry (MS), which provides a better sensitivity and specificity than traditional TLC methods. -
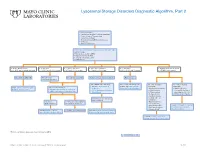
Lysosomal Storage Disorders Diagnostic Algorithm, Part 2
Lysosomal Storage Disorders Diagnostic Algorithm, Part 2 Clinical information: ■ Developmental delay/Cognitive impairment ■ Coarse features/Organomegaly ■ Dysostosis multiplex ■ Neurodegeneration/Behavioral changes ■ Ichthyosis ■ Hearing defects/loss LSDS / Lysosomal Storage Disorders Screen, Random, Urine Testing includes: ■ Mucopolysaccharides (MPS) ■ Oligosaccharides (OLIGO) ■ Ceramide trihexosides (CT) ■ Sulfatides (S) ■ OLIGO: ML II/III profile ■ S: abnormal ■ CT and S: abnormal ■ MPS and S: abnormal ■ CT: abnormal ■ OLIGO: characteristic profile ■ CT, MPS and S: normal/abnormal ■ CT, MPS and OLIGO: normal ■ MPS and OLIGO: normal ■ CT and OLIGO: normal ■ MPS, OLIGO and S: normal ■ CT, MPS and S; normal Mucolipidosis (ML) II/III Metachromatic Prosaposin/ SaposinB Multiple sulfatase deficiency (MSD) Fabry disease leukodystrophy (MLD) Order BOTH of the following: For recommended diagnostic One of the following One of the following ■ ARSAW / Arylsulfatase A, workup, see Fabry Disease suspected: suspected: Order: GNPTZ / GNPTAB Gene, Order 1 of the following: Leukocytes Diagnostic Testing Algorithm ■ Aspartylglucosaminuria ■ NGLY1 deficiency Full Gene Analysis, Varies ■ ARSAW / Arylsulfatase A, Leukocytes ■ I2SW / Iduronate-2-Sulfatase, ■ α-Mannosidosis (Congenital disorder of ■ ARSU / Arylsulfatase A, 24 Hour, Urine Whole Blood ■ β-Mannosidosis glycosylation: CDG-Iv) ■ Pompe disease ■ MOGS-CDG (CDG-IIb) ■ Sandhoff disease ■ Schindler disease If both results are deficient, ■ Sialidosis MSD confirmed If deficient, If normal, possible Saposin -
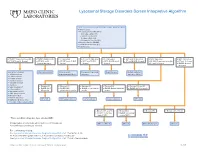
Lysosomal Storage Disorders Screen Interpretive Algorithm
Lysosomal Storage Disorders Screen Interpretive Algorithm LSDS / Lysosomal Storage Disorders Screen, Random, Urine Testing includes: ■ Mucopolysaccharides (MPS): – Dermatan sulfate (DS) – Heparan sulfate (HS) – Keratan sulfate (KS) – Chondroitin 6-sulfate (CS) ■ Oligosaccharides (OLIGO) ■ Ceramide trihexosides (CT) ■ Sulfatides (S) ■ OLIGO: Characteristic profile ■ OLIGO: MLII/III profile ■ S: Abnormal ■ CT and S: Abnormal ■ CT: Abnormal ■ MPS and S: Abnormal ■ MPS: Abnormal ■ MPS: Abnormal ■ CT, MPS and S: Normal ■ CT, MPS and S: ■ CT, MPS and OLIGO: ■ MPS and OLIGO: ■ MPS, OLIGO and S: ■ CT and OLIGO: Normal ■ OLIGO: Characteristic profile ■ CT and S: Normal Normal/abnormal Normal Normal Normal ■ CT and S: Normal ■ OLIGO: Normal/ abnormal One of the following: Mucolipidosis II/III Metachromatic Prosaposin/SaposinB Fabry Disease Multiple sulfatase ■ α-Mannosidosis leukodystrophy (MLD) deficiency deficiency (MSD) ■ β-Mannosidosis ■ Pompe disease ■ Sandhoff disease ■ Schindler disease ■ Sialidosis ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS and CS ■ Galactosialidosis* ■ OLIGO: MPS ■ OLIGO: GM1 ■ OLIGO: α-Fucosidosis ■ OLIGO: Galactosialidosis ■ OLIGO: MPS IVA profile ■ α-Fucosidosis* IVB profile gangliosidosis profile profile profile ■ Mucolipidosis II/III* ■ GM1 gangliosidosis* ■ Morquio A & B* ■ NGYL1 deficiency MPS IVB GM1 gangliosidosis -Fucosidosis Galactosialidosis MPS IVA ■ MOGS-CDG (Congenital α Disorder of Glycosylation-IIb) ■ Elevated DS and HS ■ Elevated DS ■ Elevated HS ■ Elevated DS, HS, CS ■ OLIGO: -

Lysosomal Storage Disease Panel by Next-Generation Sequencing
TEST ID: LSDP LYSOSOMAL STORAGE DISEASE PANEL BY NEXT-GENERATION SEQUENCING USEFUL FOR REFERENCE VALUES } Follow up for abnormal biochemical results and confirmation of suspected lysosomal storage An interpretive report will be disease (LSD) provided. } Identifying mutations within genes known to be associated with lysosomal storage disease, allowing for predictive testing of at-risk family members ANALYTIC TIME 4 weeks CLINICAL INFORMATION Lysosomal storage diseases (LSDs) encompass a group of over 40 inherited biochemical diseases in which genetic mutations cause defective lysosomal functioning. Lysosomes perform catabolic functions for cells, which is accomplished through activity of various proteins such as lysosomal enzymes, transport proteins, and other proteins. Functional deficits in these proteins cause an accumulation of substrates in cells leading to progressive organ dysfunction. This leads to variable clinical features that can affect the cardiovascular, neurological, ocular, and skeletal systems, among others. Clinical features are dependent on the amount and location of the substrate accumulation, but may include the following: characteristic facial features (coarse features), hepatomegaly, deafness, vision loss, abnormal skeletal findings, hydrops fetalis, ataxia, hypotonia, developmental delay/regression, and intellectual disability. Age of onset is variable, with symptoms presenting from the prenatal period to adulthood, but generally LSDs are progressive and cause significant morbidity and mortality with a decreased lifespan. Enzyme replacement therapy and oral substrate inhibitors are therapeutic options for some LSDs. LSDs are inherited in an autosomal recessive manner with the exception of Hunter, Fabry, and Danon diseases, which are X-linked. There are some founder mutations associated with particular LSDs in the Ashkenazi Jewish and Finnish populations, leading to an increased carrier frequency for some. -
The Role of Mutations on NAGA Gene in Schindler Syndrome
Mini Review Journal of Cancer Science & Therapy Volume 12:9, 2020 DOI: 10.37421/jcst.2020.12.645 ISSN: 2165-7920 Open Access The Role of Mutations on NAGA Gene in Schindler Syndrome Shahin Asadi1* and Mohaddeseh Mohsenifar2 1Division of Medical Genetics and Molecular Pathology Research, Boston Children's Hospital, Harvard University, USA 2Division of Medical Genetics and Molecular Optogenetic Research, Massachusetts Institute of Technology, USA Abstract Schindler syndrome is an inherited genetic disorder that mainly causes neurological problems. Schindler's syndrome is caused by a mutation in the NAGA gene, which is located in the long arm of chromosome 22 as 22q13.2. Amniocentesis or chorionic villus sampling can be used to screen for the disease before birth. After birth, urine tests, along with blood tests and skin biopsies can be used to diagnose Schindler disease. Genetic testing is also always an option, since different forms of Schindler disease have been mapped to the same gene on chromosome 22; though different changes (mutations) of this gene are responsible for the infantile- and adult-onset forms of the disease. The Genetic testing Registry can be used to acquire information about the genetic tests for this condition. Keywords: Schindler syndrome • Genetic disorder • NAGA gene • Kanzaki disease Schindler's syndrome type 2 is also known as Kanzaki disease, which Introduction is milder than type 1 syndrome and is most commonly seen in adulthood. Affected individuals may develop mild cognitive impairment and hearing loss Schindler disease, also known as Kanzaki disease and alpha-N- due to inner ear abnormalities (sensory hearing loss). They may experience acetylgalactosaminidase deficiency is a rare disease found in humans. -
Neurodegenerative Conditions of Ophthalmic Importance Mark S
10 Neurodegenerative Conditions of Ophthalmic Importance Mark S. Borchert and Sarah Ying he pediatric ophthalmologist is frequently asked to evaluate Tchildren with multiple neurological impairments for oph- thalmic manifestations of heritable neurological diseases. Occa- sionally these are diseases with specific ocular findings such as Kayser–Fleischer rings in Wilson’s disease or cherry-red spots in Tay–Sachs disease. More typically, they are diseases with non- specific findings such as optic atrophy or nystagmus. Such find- ings can be used to help direct the neurological workup. Less commonly, these children present to the ophthalmologist with ocular complaints primarily. The ophthalmologist must recog- nize those signs and symptoms that may be associated with neu- rodegenerative conditions and institute an appropriate workup. Although all these conditions are rare, a general ophthalmologist is likely to encounter several of them in their career. Most of the neurodegenerative conditions are metabolic and represent dysfunction of specific enzymes or enzyme systems. Many of these enzyme systems are associated with particular cellular organelles, allowing for the classification of these dis- eases according to the organelle that is disrupted. Examples of these are the lysosomal storage diseases, the mitochondrial dis- orders, and the peroxisomal diseases. Other neurodegenerative diseases have characteristic clinical and pathological findings without known enzyme defects. Clinically, these diseases each have their own stereotypical time and mode of onset, which allows for easy division of the neurodegenerative conditions into groups that present in infancy, early childhood, or late childhood (Tables 10-1 through 10-3). 324 chapter 10: neurodegenerative conditions 325 LYSOSOMAL STORAGE DISEASES AND LEUKODYSTROPHIES The lysosomal storage diseases are caused by dysfunction of lysosomal enzymes involved in the catabolism of complex lipids and sugars normally present in tissues, resulting in accumula- tion of intracellular storage products. -

Metabolic Disorders (Children)
E06/S/b 2013/14 NHS STANDARD CONTRACT METABOLIC DISORDERS (CHILDREN) PARTICULARS, SCHEDULE 2 – THE SERVICES, A – SERVICE SPECIFICATION Service Specification E06/S/b No. Service Metabolic Disorders (Children) Commissioner Lead Provider Lead Period 12 months Date of Review 1. Population Needs 1.1 National/local context and evidence base National Context Inherited Metabolic Disorders (IMDs) cover a group of over 600 individual conditions, each caused by defective activity in a single enzyme or transport protein. Although individually metabolic conditions are rare, the incidence being less than 1.5 per 10,000 births, collectively they are a considerable cause of morbidity and mortality. The diverse range of conditions varies widely in presentation and management according to which body systems are affected. For some patients presentation may be in the newborn period, whereas for others with the same disease (but a different genetic mutation) onset may be later, including adulthood. Without early identification and/or introduction of specialist diet or drug treatments, patients face severe disruption of metabolic processes in the body such as energy production, manufacture of breakdown of proteins, and management and storage of fats and fatty acids. The result is that patients have either a deficiency of products essential to health or an accumulation of unwanted or toxic products. Without treatment many conditions can lead to severe learning or physical disability and death at an early age. The rarity and complex nature of IMD requires an integrated specialised clinical and laboratory service to provide satisfactory diagnosis and management. This is in keeping with the recommendation of the 1 NHS England E06/S/b © NHS Commissioning Board, 2013 The NHS Commissioning Board is now known as NHS England Department of Health’s UK Plan for Rare Disorders consultation to use specialist centres Approximately 10-12,000 paediatric and adult patients attend UK specialist IMD centres, but a significant number of patients remain undiagnosed or are ‘lost to follow-up’. -

Glycoprotein Lysosomal Storage Disorders: K- and L-Mannosidosis, Fucosidosis and K-N-Acetylgalactosaminidase De¢Ciency
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1455 (1999) 69^84 www.elsevier.com/locate/bba Review Glycoprotein lysosomal storage disorders: K- and L-mannosidosis, fucosidosis and K-N-acetylgalactosaminidase de¢ciency Jean-Claude Michalski a;*, Andre¨ Klein b a Laboratoire de Chimie Biologique, UMR 8576 CNRS (UMR 111 CNRS), Universite¨ des Sciences et Technologies de Lille, 59655 Villeneuve d'Ascq Cedex, France b Unite¨ 377 INSERM, C.H.U., Place de Verdun, 59045 Lille Cedex, France Received 22 February 1999; received in revised form 21 June 1999; accepted 21 June 1999 Abstract Glycoproteinoses belong to the lysosomal storage disorders group. The common feature of these diseases is the deficiency of a lysosomal protein that is part of glycan catabolism. Most of the lysosomal enzymes involved in the hydrolysis of glycoprotein carbohydrate chains are exo-glycosidases, which stepwise remove terminal monosaccharides. Thus, the deficiency of a single enzyme causes the blockage of the entire pathway and induces a storage of incompletely degraded substances inside the lysosome. Different mutations may be observed in a single disease and in all cases account for the non- expression of lysosomal glycosidase activity. Different clinical phenotypes generally characterize a specific disorder, which rather must be described as a continuum in severity, suggesting that other biochemical or environmental factors influence the course of the disease. This review provides details on clinical features, genotype-phenotype correlations, enzymology and biochemical storage of four human glycoprotein lysosomal storage disorders, respectively K- and L-mannosidosis, fucosidosis and K-N-acetylgalactosaminidase deficiency.