Neurodegenerative Conditions of Ophthalmic Importance Mark S
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Dystonia and Chorea in Acquired Systemic Disorders
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.65.4.436 on 1 October 1998. Downloaded from 436 J Neurol Neurosurg Psychiatry 1998;65:436–445 NEUROLOGY AND MEDICINE Dystonia and chorea in acquired systemic disorders Jina L Janavs, Michael J AminoV Dystonia and chorea are uncommon abnormal Associated neurotransmitter abnormalities in- movements which can be seen in a wide array clude deficient striatal GABA-ergic function of disorders. One quarter of dystonias and and striatal cholinergic interneuron activity, essentially all choreas are symptomatic or and dopaminergic hyperactivity in the nigros- secondary, the underlying cause being an iden- triatal pathway. Dystonia has been correlated tifiable neurodegenerative disorder, hereditary with lesions of the contralateral putamen, metabolic defect, or acquired systemic medical external globus pallidus, posterior and poste- disorder. Dystonia and chorea associated with rior lateral thalamus, red nucleus, or subtha- neurodegenerative or heritable metabolic dis- lamic nucleus, or a combination of these struc- orders have been reviewed frequently.1 Here we tures. The result is decreased activity in the review the underlying pathogenesis of chorea pathways from the medial pallidus to the and dystonia in acquired general medical ventral anterior and ventrolateral thalamus, disorders (table 1), and discuss diagnostic and and from the substantia nigra reticulata to the therapeutic approaches. The most common brainstem, culminating in cortical disinhibi- aetiologies are hypoxia-ischaemia and tion. Altered sensory input from the periphery 2–4 may also produce cortical motor overactivity medications. Infections and autoimmune 8 and metabolic disorders are less frequent and dystonia in some cases. To date, the causes. Not uncommonly, a given systemic dis- changes found in striatal neurotransmitter order may induce more than one type of dyski- concentrations in dystonia include an increase nesia by more than one mechanism. -

Fingolimod Rescues Demyelination in a Mouse Model of Krabbe's Disease
Research Articles: Neurobiology of Disease Fingolimod rescues demyelination in a mouse model of Krabbe's disease https://doi.org/10.1523/JNEUROSCI.2346-19.2020 Cite as: J. Neurosci 2020; 10.1523/JNEUROSCI.2346-19.2020 Received: 30 September 2019 Revised: 17 December 2019 Accepted: 21 January 2020 This Early Release article has been peer-reviewed and accepted, but has not been through the composition and copyediting processes. The final version may differ slightly in style or formatting and will contain links to any extended data. Alerts: Sign up at www.jneurosci.org/alerts to receive customized email alerts when the fully formatted version of this article is published. Copyright © 2020 Be´chet et al. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International license, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed. 1 Fingolimod rescues demyelination in a mouse model of Krabbe’s disease 2 Sibylle Béchet, Sinead O’Sullivan, Justin Yssel, Steven G. Fagan, Kumlesh K. Dev 3 Drug Development, School of Medicine, Trinity College Dublin, IRELAND 4 5 Corresponding author: Prof. Kumlesh K. Dev 6 Corresponding author’s address: Drug Development, School of Medicine, Trinity College 7 Dublin, IRELAND, D02 R590 8 Corresponding author’s phone and fax: Tel: +353 1 896 4180 9 Corresponding author’s e-mail address: [email protected] 10 11 Number of pages: 35 pages 12 Number of figures: 9 figures 13 Number of words (Abstract): 216 words 14 Number of words (Introduction): 641 words 15 Number of words (Discussion): 1475 words 16 17 Abbreviated title: Investigating the use of FTY720 in Krabbe’s disease 18 19 Acknowledgement statement (including conflict of interest and funding sources): This work 20 was supported, in part, by Trinity College Dublin, Ireland and the Health Research Board, 21 Ireland. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

Post Stroke Focal Aware Seizures Presenting As Delayed Onset Choreoathetosis
Lehigh Valley Health Network LVHN Scholarly Works Department of Medicine Post Stroke Focal Aware Seizures Presenting as Delayed Onset Choreoathetosis Artish Patel USF MCOM - LVHN Campus, [email protected] Patrick Davis USF MCOM - LVHN Campus, [email protected] Beth Stepanczuk MD Lehigh Valley Health Network, [email protected] Follow this and additional works at: https://scholarlyworks.lvhn.org/medicine Part of the Medicine and Health Sciences Commons Published In/Presented At Patel, A., Davis, P., & Stepanczuk, B. (2021, February 9-13). Post Stroke Focal Aware Seizures Presenting as Delayed Onset Choreoathetosis. [Poster presentation]. Association of Academic Physiatrists Annual Meeting, Virtual. This Poster is brought to you for free and open access by LVHN Scholarly Works. It has been accepted for inclusion in LVHN Scholarly Works by an authorized administrator. For more information, please contact [email protected]. Post Stroke Focal Aware Seizures Presenting as Delayed Onset Choreoathetosis Artish Patel, MD, BS, Patrick Davis, MD, BS, and Beth Stepanczuk, MD Lehigh Valley Health Network; Allentown, Pa. Setting Case Description Discussion Outpatient follow-up office She initially presented with acute onset confusion, nausea, This patient offered a rare case of focal aware seizures vomiting, and aphasia. Imaging revealed acute left parietal presenting as delayed onset choreoathetosis of the right Patient temporal intraparenchymal hemorrhage with surrounding upper extremity secondary to acute left parietal temporal 39-year-old female with history of migraines and recent left parietal edema and restricted diffusion consistent with infarction intraparenchymal hemorrhage. It has been documented that temporal intraparenchymal hemorrhage presenting at three- secondary to superior sagittal sinus thrombosis. -

Paraneoplastic Neurological and Muscular Syndromes
Paraneoplastic neurological and muscular syndromes Short compendium Version 4.5, April 2016 By Finn E. Somnier, M.D., D.Sc. (Med.), copyright ® Department of Autoimmunology and Biomarkers, Statens Serum Institut, Copenhagen, Denmark 30/01/2016, Copyright, Finn E. Somnier, MD., D.S. (Med.) Table of contents PARANEOPLASTIC NEUROLOGICAL SYNDROMES .................................................... 4 DEFINITION, SPECIAL FEATURES, IMMUNE MECHANISMS ................................................................ 4 SHORT INTRODUCTION TO THE IMMUNE SYSTEM .................................................. 7 DIAGNOSTIC STRATEGY ..................................................................................................... 12 THERAPEUTIC CONSIDERATIONS .................................................................................. 18 SYNDROMES OF THE CENTRAL NERVOUS SYSTEM ................................................ 22 MORVAN’S FIBRILLARY CHOREA ................................................................................................ 22 PARANEOPLASTIC CEREBELLAR DEGENERATION (PCD) ...................................................... 24 Anti-Hu syndrome .................................................................................................................. 25 Anti-Yo syndrome ................................................................................................................... 26 Anti-CV2 / CRMP5 syndrome ............................................................................................ -

Adult-Onset Krabbe Disease in Two Generations of a Chinese Family
174 Original Article on Translational Neurodegeneration Page 1 of 6 Adult-onset Krabbe disease in two generations of a Chinese family Tongxia Zhang1, Chuanzhu Yan1,2,3, Kunqian Ji1, Pengfei Lin1, Lingyi Chi2,4, Xiuhe Zhao1, Yuying Zhao1 1Research Institute of Neuromuscular and Neurodegenerative Diseases and Department of Neurology, 2Brain Science Research Institute, Qilu Hospital, Shandong University, Jinan 250012, China; 3Mitochondrial Medicine Laboratory, Qilu Hospital (Qingdao), Shandong University, Qingdao 266035, China; 4Department of Neurosurgery, Qilu Hospital, Shandong University, Jinan 250012, China Contributions: Conception and design: Y Zhao, C Yan; (II) Administrative support: C Yan; (III) Provision of study materials or patients: Y Zhao, T Zhang; (IV) Collection and assembly of data: T Zhang, K Ji, P Lin, L Chi, X Zhao; (V) Data analysis and interpretation: T Zhang, K Ji, P Lin, L Chi, X Zhao; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Dr. Yuying Zhao. Research Institute of Neuromuscular and Neurodegenerative Diseases and Department of Neurology, Qilu Hospital, Shandong University, Jinan 250012, China. Email: [email protected]. Background: Krabbe disease (KD) is a rare autosomal recessive lysosomal storage disorder caused by deficiency of the galactocerebrosidase (GALC) enzyme. The adult-onset KD is infrequent, and often presenting with slowly progressive spastic paraplegia. Herein, we describe a two-generation concomitant Chinese pedigree of adult-onset KD in which the proband presented with acute hemiplegia at onset. Methods: We collected the clinical and neuroimaging data of the pedigree. GALC enzyme activity detection and gene analysis were performed to confirm the diagnosis. Moreover, we reviewed all studies available on PubMed to understand the correlationship between phenotype and genotype of the identified mutations. -

EMERGENCY CARE AID for ALTERNATING HEMIPLEGIA of CHILDHOOD (AHC) PATIENTS a Reference Guide for Medical Professionals
EMERGENCY CARE AID FOR ALTERNATING HEMIPLEGIA OF CHILDHOOD (AHC) PATIENTS A Reference Guide for Medical Professionals AHC DEFINITION: (Handb Clin Neurol. 2013; 112:821-6) Alternating hemiplegia of childhood (AHC) is a very rare disease characterized by recurrent attacks of loss of muscular tone resulting in hypomobility of one side of the body. The etiology of the disease is due to ATP1A3 gene mutations in the majority of patients. AHC has an onset in the first few months of life. Hemiplegic episodes are often accompanied by other paroxysmal manifestations, such as lateral eyes and head deviation toward the hemiplegic side and a very peculiar monocular nystagmus. Movement disorders such as dystonia and abnormal movements are frequent. Cognitive delay of variable degree is a common feature. Epilepsy has been reported in 50% of the cases, but seizure onset is usually during the third or fourth year of life. Many drugs have been used in AHC with very few results. Flunarizine has the most supportive anecdotal evidence regarding efficacy. AHC RISK FACTORS: TREATMENT: (Neurol Genet. 2017 Apr; 3(2): e139) There is no specific treatment for AHC. There is no approved medication or device that alters the underlying deficit in the Na+/K+ pump (see Flunarizine next page). NEUROLOGICAL: (Neurology. 2019 Sep 24;93(13)) Epilepsy is present in over 50% of AHC patients. Epilepsy in AHC can be focal or generalized seizures. Dystonia is also a common feature that affects the majority of patients dealing with AHC. Patients with AHC can deteriorate either abruptly or gradually at the motor and intellectual function level, depending on the severity of their disease. -
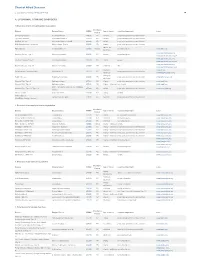
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none -

2021 Jp Morgan Healthcare Conference
Cover page, paste image over entire page 2021 J.P. MORGAN HEALTHCARE CONFERENCE: DAY 1 January 2021|1 Summary The 39th annual J.P. Morgan Healthcare Conference (JPM) is being held virtually from January 11-14, 2021. A list of events and catalysts that were announced or updated at the conference today is included in this report. Below are some key points from today’s company presentations. Key Takeaways - Day 1 Mega Cap Companies • Amgen (AMGN) Chairman and CEO, Robert Bradway, started his presentation by focusing on Amgen’s two late stage assets, sotorasib and tezepelumab, at the J.P. Morgan Healthcare Conference given their large impact events expected this year, their potential to be first-in-class therapies, and the large unmet need they would be addressing in their indications. Key data catalysts for sotorasib in 2021 include a release of full Phase II results in KRAS G12C- mutant advanced NSCLC patients on January 29th at the World Lung Conference, Phase II colorectal cancer data in the first half of 2021, and initial data on several combinations with the KRAS inhibitor also in the first half of 2021. Regulatory submissions for sotorasib have been completed in the US and EU, and management indicates they are accelerating global launch preparations for sotorasib in anticipation of projected approvals this year. Successful approval would provide an effective targeted therapy option for previously treated NSCLC patients who have KRAS G12C-mutated locally advanced or metastatic, which account for 13% of the NSCLC patient population. In the long term, the Company indicates a possible exploration of label expansions to earlier lines of therapy and/or combinations with other therapies. -
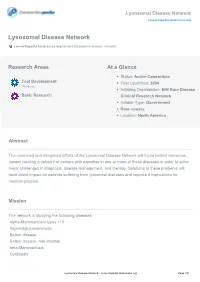
Lysosomal Disease Network Consortiapedia.Fastercures.Org
Lysosomal Disease Network consortiapedia.fastercures.org Lysosomal Disease Network consortiapedia.fastercures.org/consortia/lysosomal-disease-network/ Research Areas At a Glance Status: Active Consortium Tool Development Year Launched: 2004 Resource Initiating Organization: NIH Rare Disease Basic Research Clinical Research Network Initiator Type: Government Rare disease Location: North America Abstract The combined and integrated efforts of the Lysosomal Disease Network will focus limited resources toward creating a network of centers with expertise in one or more of these diseases in order to solve major challenges in diagnosis, disease management, and therapy. Solutions to these problems will have direct impact on patients suffering from lysosomal diseases and important implications for medical practice. Mission The network is studying the following diseases: alpha-Mannosidosis types I / II Aspartylglucosaminuria Batten disease Batten disease, late infantile beta-Mannosidosis Cystinosis Lysosomal Disease Network - consortiapedia.fastercures.org Page 1/5 Lysosomal Disease Network consortiapedia.fastercures.org Danon disease Fabry disease Farber disease Fucosidosis Galactosialidosis types I / II Gaucher disease GM1-Gangliosidosis types I/II/III GM2-Gangliosidosis Hunter syndrome Hurler syndrome I-cell disease Krabbe disease Maroteaux-Lamy syndrome Metachromatic leukodystrophy Morquio syndrome Mucolipidosis type IV Mucopolysaccharidosis type IX Multiple sulfatase deficiency Niemann-Pick disease Northern Epilepsy Pompe disease pseudo-Hurler -

Pathologic Classification of White Matter Disorders • Demyelinating
Pathologic classification of white matter disorders • Demyelinating - loss of normal myelin autoimmune/inflammatory component • Dysmyelinating - loss of chemically abnormal myelin •Hypomyelinating - paucity of myelin formation • Myelinolytic (Spongiform) - cytotoxic loss of myelin, intramyelinolytic edema Oligodendrocytes make myelin in the CNS Multiple axons are myelininated by one oligodendrocyte 1 Demyelinating Diseases • Multiple Sclerosis • Acute Disseminated Encephalomyelitis • Acute Hemorrhagic Leukoencephalitis • Progressive Multifocal Leukoencephalopathy • Subacute Sclerosing Panencephalitis • Idopathic Polyneuritis (Landry-Guillain-Barre) Multiple Sclerosis • Episodic neurologic signs and symptoms referable to different parts of the neuraxis (“disseminated in time and space”) • Attacks followed by complete or partial remission • Peak age of onset is 20-40 years; more common in women • Chronic relapsing (“classical”) and rapidly progressing forms • Diagnosis established by clinical history, MRI, CSF analysis (oligoclonal bands) 2 “Classical” Multiple Sclerosis • Prevalence: • 30-120/100,000 in Northern Latitudes • Etiology: • Genetic Factors • Environmental Factors • Immunologic Pathogenesis Multiple Sclerosis - CT Scans Complete/partial resolution Neurologic symptoms present of neurologic symptoms 3 Multiple Sclerosis Plaque - periventricular 4 Shadow Plaque = Partial remyelination H&E Luxol Fast Blue MS plaques may involve “gray matter” regions (e.g. deep nuclei in forebrain, brain stem) where there is a close mixture of myelinated -

Early Progression of Krabbe Disease in Patients with Symptom Onset Between 0 and 5 Months Maria L
Beltran-Quintero et al. Orphanet Journal of Rare Diseases (2019) 14:46 https://doi.org/10.1186/s13023-019-1018-4 RESEARCH Open Access Early progression of Krabbe disease in patients with symptom onset between 0 and 5 months Maria L. Beltran-Quintero1†, Nicholas A. Bascou1†, Michele D. Poe1, David A. Wenger2, Carlos A. Saavedra-Matiz3, Matthew J. Nichols3 and Maria L. Escolar1* Abstract Background: Krabbe disease is a rare neurological disorder caused by a deficiency in the lysosomal enzyme, β- galactocerebrosidase, resulting in demyelination of the central and peripheral nervous systems. If left without treatment, Krabbe disease results in progressive neurodegeneration with reduced quality of life and early death. The purpose of this prospective study was to describe the natural progression of early onset Krabbe disease in a large cohort of patients. Methods: Patients with early onset Krabbe disease were prospectively evaluated between 1999 and 2018. Data sources included diagnostic testing, parent questionnaires, standardized multidisciplinary neurodevelopmental assessments, and neuroradiological and neurophysiological tests. Results: We evaluated 88 children with onset between 0 and 5 months. Median age of symptom onset was 4 months; median time to diagnosis after onset was 3 months. The most common initial symptoms were irritability, feeding difficulties, appendicular spasticity, and developmental delay. Other prevalent symptoms included axial hypotonia, abnormal deep tendon reflexes, constipation, abnormal pupillary response, scoliosis, loss of head control, and dysautonomia. Results of nerve conduction studies showed that 100% of patients developed peripheral neuropathy by 6 months of age. Median galactocerebrosidase enzyme activity was 0.05 nmol/h/mg protein. The median survival was 2 years.