Schindler Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 8,323,640 B2 Sakuraba Et Al
USOO8323640B2 (12) United States Patent (10) Patent No.: US 8,323,640 B2 Sakuraba et al. (45) Date of Patent: *Dec. 4, 2012 (54) HIGHLY FUNCTIONAL ENZYME HAVING OTHER PUBLICATIONS O-GALACTOSIDASE ACTIVITY Broun et al., Catalytic plasticity of fatty acid modification enzymes (75) Inventors: Hitoshi Sakuraba, Abiko (JP); Youichi underlying chemical diversity of plant lipids. Science, 1998, vol. 282: Tajima, Tokyo (JP); Mai Ito, Tokyo 1315-1317. (JP); Seiichi Aikawa, Tokyo (JP); Cameron ER. Recent advances in transgenic technology. 1997, vol. T: 253-265. Fumiko Aikawa, Tokyo (JP) Chica et al., Semi-rational approaches to engineering enzyme activ (73) Assignees: Tokyo Metropolitan Organization For ity: combining the benefits of directed evolution and rational design. Curr, opi. Biotechnol., 2005, vol. 16:378-384. Medical Research, Tokyo (JP); Altif Couzin et al. As Gelsinger case ends, Gene therapy Suffers another Laboratories, Tokyo (JP) blow. Science, 2005, vol. 307: 1028. Devos et al., Practical Limits of Function prediction. Proteins: Struc (*) Notice: Subject to any disclaimer, the term of this ture, Function, and Genetics. 2000, vol. 41: 98-107. patent is extended or adjusted under 35 Donsante et al., AAV vector integration sites in mouse hapatocellular U.S.C. 154(b) by 0 days. carcinoma. Science, 2007. vol. 317: 477. Juengst ET., What next for human gene therapy? BMJ., 2003, vol. This patent is Subject to a terminal dis 326: 1410-1411. claimer. Kappel et al., Regulating gene expression in transgenic animals. Current Opinion in Biotechnology 1992, vol. 3: 548-553. (21) Appl. No.: 13/052,632 Kimmelman J. Recent developments in gene transfer: risk and eth ics. -

Supplementary Information Biocementation of Soil by Calcite/Aragonite Precipitation Using Pseudomonas Azotoformans and Citrobact
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supplementary Information Biocementation of soil by calcite/aragonite precipitation using Pseudomonas azotoformans and Citrobacter freundii derived enzymes Heba Abdel-Aleem1, Tarek Dishisha1, Amal Saafan2, Abduallah A. AbouKhadra3 and Yasser Gaber1,4,* 1Department of Microbiology and Immunology, Faculty of Pharmacy, Beni-Suef University, 62511, Beni-Suef, Egypt 2Department of Pharmaceutical Microbiology, Faculty of Pharmacy, Menoufia university, Shebeen El-kom, Egypt 3Department of Civil Engineering, Facutly of Engineering, Beni-Suef University, 62511, Beni- Suef, Egypt 4Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Mutah University, Al-karak, 61710, Jordan *Corresponding author: E-mail: [email protected] Tel. int +20 82 216 2133 ; Fax. int +20 82 216 2133 1 Supplementary Table 1: strains identified using BCL card (Gram positive spore forming bacilli) Sample/test A1 B1 B2 Y1a Y2a BXYL - - - - - BXYL (betaxylosidase), LysA - - - - - LysA (L- lysine arylamidase), AspA - - - + - AspA (L aspartate arylamidase), LeuA + + + + + LeuA (leucine arylamidase), PheA - + - - - PheA (phenylalanine arylamidase), ProA - - - + - ProA (L-proline arylamidase), BGAL - - - - - BGAL (beta galactosidase), PyrA + + + + + PyrA (L-pyrrolydonyl arylamidase), AGAL - - - - - AGAL (alpha galactosidase), AlaA - - - + - AlaA (alanine arylamidase), TyrA - - - - - TyrA (tyrosine arylamidase), BNAG + + + + + BNAG (beta-N-acetyl -

Base Excision Repair Deficient Mice Lacking the Aag Alkyladenine DNA Glycosylase
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 13087–13092, November 1997 Genetics Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase BEVIN P. ENGELWARD*†,GEERT WEEDA*‡,MICHAEL D. WYATT†,JOSE´ L. M. BROEKHOF‡,JAN DE WIT‡, INGRID DONKER‡,JAMES M. ALLAN†,BARRY GOLD§,JAN H. J. HOEIJMAKERS‡, AND LEONA D. SAMSON†¶ †Department of Molecular and Cellular Toxicology, Harvard School of Public Health, 665 Huntington Avenue, Boston, MA 02115; ‡Department of Cell Biology and Genetics, Medical Genetics Centre, Erasmus University, P.O. Box 1738, 3000 DR, Rotterdam, The Netherlands; and §Eppley Institute for Research in Cancer and Allied Diseases and Department of Pharmaceutical Sciences, University of Nebraska Medical Center, Omaha, NE 68198-6805 Edited by Philip Hanawalt, Stanford University, Stanford, CA, and approved September 30, 1997 (received for review August 8, 1997) ABSTRACT 3-methyladenine (3MeA) DNA glycosylases 11). The precise biological effects of all of the DNA lesions remove 3MeAs from alkylated DNA to initiate the base repaired by mammalian 3MeA DNA glycosylases are not yet excision repair pathway. Here we report the generation of mice known for mammals, though there is strong evidence that deficient in the 3MeA DNA glycosylase encoded by the Aag 3MeA is cytotoxic (12), and other lesions may be mutagenic, (Mpg) gene. Alkyladenine DNA glycosylase turns out to be the namely Hx (13), 8oxoG (14), 1,N6-ethenoadenine («A), and major DNA glycosylase not only for the cytotoxic 3MeA DNA N2,3-ethenoguanine (15, 16). It is important to note that each lesion, but also for the mutagenic 1,N6-ethenoadenine («A) of these DNA lesions can arise spontaneously in the DNA of and hypoxanthine lesions. -

Targeted Deletion of Alkylpurine-DNA-N-Glycosylase in Mice Eliminates Repair of 1,N6-Ethenoadenine and Hypoxanthine but Not of 3,N4-Ethenocytosine Or 8-Oxoguanine
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 12869–12874, November 1997 Biochemistry Targeted deletion of alkylpurine-DNA-N-glycosylase in mice eliminates repair of 1,N6-ethenoadenine and hypoxanthine but not of 3,N4-ethenocytosine or 8-oxoguanine B. HANG*, B. SINGER*†,G.P.MARGISON‡, AND R. H. ELDER‡ *Donner Laboratory, Lawrence Berkeley National Laboratory, University of California, Berkeley, CA 94720; and ‡Cancer Research Campaign Section of Genome Damage and Repair, Paterson Institute for Cancer Research, Christie Hospital (National Health Service) Trust, Manchester, M20 4BX, United Kingdom Communicated by H. Fraenkel-Conrat, University of California, Berkeley, CA, October 2, 1997 (received for review September 18, 1997) ABSTRACT It has previously been reported that 1,N6- A separate line of experimentation to assess the in vivo ethenoadenine («A), deaminated adenine (hypoxanthine, Hx), mutagenicity of various modified bases in differing repair and 7,8-dihydro-8-oxoguanine (8-oxoG), but not 3,N4- backgrounds used defined synthetic oligonucleotides or glo- ethenocytosine («C), are released from DNA in vitro by the bally modified vectors (12–15). Related to this are numerous DNA repair enzyme alkylpurine-DNA-N-glycosylase (APNG). in vitro experiments also using site-directed mutagenesis in To assess the potential contribution of APNG to the repair of which replication efficiency and changed base pairing could be each of these mutagenic lesions in vivo, we have used cell-free quantitated [reviewed by Singer and Essigmann, (15)]. These extracts of tissues from APNG-null mutant mice and wild-type data all yielded basic information on the potential biological controls. The ability of these extracts to cleave defined oli- role of modified bases in the absence of any repair capacity. -

Active Hydrogen Bond Network (AHBN) and Applications for Improvement of Thermal Stability and Ph-Sensitivity of Pullulanase from Bacillus Naganoensis
RESEARCH ARTICLE Active Hydrogen Bond Network (AHBN) and Applications for Improvement of Thermal Stability and pH-Sensitivity of Pullulanase from Bacillus naganoensis Qing-Yan Wang1☯, Neng-Zhong Xie1☯, Qi-Shi Du1,2*, Yan Qin1, Jian-Xiu Li1,3, Jian- Zong Meng3, Ri-Bo Huang1,3 a1111111111 1 State Key Laboratory of Biomass Enzyme Technology, National Engineering Research Center for Non- Food Biorefinery, Guangxi Academy of Sciences, Nanning, Guangxi, China, 2 Gordon Life Science Institute, a1111111111 Belmont, MA, United States of America, 3 Life Science and Technology College, Guangxi University, a1111111111 Nanning, Guangxi, China a1111111111 a1111111111 ☯ These authors contributed equally to this work. * [email protected] Abstract OPEN ACCESS Citation: Wang Q-Y, Xie N-Z, Du Q-S, Qin Y, Li J-X, A method, so called ªactive hydrogen bond networkº (AHBN), is proposed for site-directed Meng J-Z, et al. (2017) Active Hydrogen Bond mutations of hydrolytic enzymes. In an enzyme the AHBN consists of the active residues, Network (AHBN) and Applications for functional residues, and conservative water molecules, which are connected by hydrogen Improvement of Thermal Stability and pH- bonds, forming a three dimensional network. In the catalysis hydrolytic reactions of hydro- Sensitivity of Pullulanase from Bacillus naganoensis. PLoS ONE 12(1): e0169080. lytic enzymes AHBN is responsible for the transportation of protons and water molecules, doi:10.1371/journal.pone.0169080 and maintaining the active and dynamic structures of enzymes. The AHBN of pullulanase Editor: Eugene A. Permyakov, Russian Academy of BNPulA324 from Bacillus naganoensis was constructed based on a homologous model Medical Sciences, RUSSIAN FEDERATION structure using Swiss Model Protein-modeling Server according to the template structure of Received: October 6, 2016 pullulanase BAPulA (2WAN). -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Partial Purification of the Mammalian Alpha- Glucosidase Inhibitor from Melothria Sp. (Fam. Cucurbitaceae) Leaves and Stem Extract: in Vitro
44 Partial Purification of the Mammalian Alpha- glucosidase Inhibitor from Melothria sp. (Fam. Cucurbitaceae) Leaves and Stem Extract: In Vitro Edmark C. Kamantigue1*, Noel S. Quiming2, Judylynn N. Solidum3, and Marilou G. Nicolas2 1 Pharmacy Department, College of Allied Health, National University, 2Department of Physical Science and Mathematics, College of Arts and Sciences, University of the Philippines Manila, and 3Pharmaceutical Chemistry Department, College of Pharmacy, University of the Philippines Manila *Corresponding Author: [email protected] Abstract: Diabetes Mellitus Type 2 (DM2) is a chronic disease characterized by insufficient insulin levels, pancreatic beta cells function loss and insulin resistance in peripheral tissue. Voglibose and acarbose are the clinically used alpha-glucosidase inhibitor; however, adverse effects such as nausea, diarrhea, and bloating hinder the utilization of these agents. Hence, there is a need to explore for an alternative drug that has fewer side effects, better activity, and more affordable than the current commercial drugs. In this study, Melothria sp. was purified using normal phase column chromatography. Seven fractions (fraction A-G) were separated and tested for mammalian alpha-glucosidase assay in vitro and phytochemical screening was conducted to determine the metabolites present in each fraction. Fraction F showed a promising activity against the enzyme and comparable to other natural products and commercial alpha- glucosidase agent, acarbose. Phenolic compounds such as tannins and flavonoids based on the phytochemical screening were detected present in the sample which can be responsible in the inhibition activity of the semi-purified extracts. Keywords: Diabetes Mellitus; Mammalian alpha-glucosidase; Piri-pipino; Cucurbitaceae; Melothria sp. 45 1. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Centometabolic® More Answers Today
CentoMetabolic® More Answers Today. More Options Tomorrow. PRODUCT SHEET CentoMetabolic® Inherited metabolic disorders (IMDs) are a group of rare conditions caused by genetic defects that disrupt the cellular metabolism. A growing number of IMDs are treatable if diagnosed early, but can be quickly fatal without prompt identification. With a multiomic approach, we can help you and your patients to accelerate the critical journey from symptoms to diagnosis by avoiding stepwise testing – saving time, resources, and pivotal years amid often rapid IMD progression. CENTOGENE’s multiomic panel – CentoMetabolic® – has been designed to test for a wide range of IMDs – integrating genetic and biochemical testing, including enzyme assays as well as a selection of proprietary biomarkers. When genetic variants relevant to your patient are detected via CentoMetabolic, we will automatically complement the analysis with biomarker and /or enzyme testing (if applicable) and include the results in your medical report. In addition, CentoMetabolic includes an evaluation of copy number variants (CNV) at no extra cost. CentoMetabolic gives you the confidence of a complete clinical picture, while laying the roadmap to personalized treatment options. The CENTOGENE Advantage • Curated to provide the most valuable information • High-quality analysis for precise clinical interpretation for diagnostic decisions, prognosis, and therapeutic using advanced bioinformatics and artificial intelligence- approaches powered tools • The most up-to-date panel gene content including -
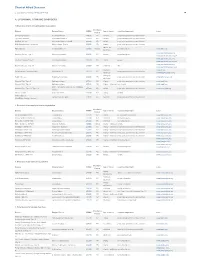
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none