Lysosomal Storage Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

File Download
Radiological and clinical characterization of the lysosomal storage disorders: non-lipid disorders Minzhi Xing, Emory University E I Parker, Emory University A Moreno-De-Luca, Geisinger Health System Elie Harmouche, Emory University Michael Terk, Emory University Journal Title: British Journal of Radiology Volume: Volume 87, Number 1033 Publisher: British Institute of Radiology | 2014-01-01, Pages 20130467-20130467 Type of Work: Article | Final Publisher PDF Publisher DOI: 10.1259/bjr.20130467 Permanent URL: https://pid.emory.edu/ark:/25593/pr2p5 Final published version: http://dx.doi.org/10.1259/bjr.20130467 Copyright information: © 2014 The Authors. This is an Open Access article distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/), which permits distribution of derivative works, making multiple copies, distribution, public display, and publicly performance, provided the original work is properly cited. This license requires credit be given to copyright holder and/or author, copyright and license notices be kept intact. Accessed September 24, 2021 3:09 PM EDT BJR © 2014 The Authors. Published by the British Institute of Radiology Received: Revised: Accepted: doi: 10.1259/bjr.20130467 28 July 2013 29 October 2013 8 November 2013 Cite this article as: Xing M, Parker EI, Moreno-De-Luca A, Harmouche E, Terk MR. Radiological and clinical characterization of the lysosomal storage disorders: non-lipid disorders. Br J Radiol 2014;87:20130467. REVIEW ARTICLE Radiological -

Lysosomal Diseases
THE GLYCOPROTEINOSES: SECOND INTERNATIONAL WORKSHOP ON ADVANCES IN PATHOGENESIS AND THERAPY JULY 26-27, 2007 ANN ARBOR, MICHIGAN Scientific Conference Day 1 Morning Sessions, July 26 7:30-8:15 Continental Breakfast 8:15-8:30 Welcome and Introductions, Opening Remarks – Goals of the conference, progress since the last conference - Steven Walkley (with additional comments by officials from ISMRD, NINDS, ORD) 8:30-9:15 Plenary Address Cross-Correction: Looking Back, Looking Forward – Elizabeth Neufeld Session 1: Animal Models, Pathogenesis, Experimental Therapies (Chair, Mark Haskins) α-Mannosidosis 9:15-9:35 Enzyme replacement therapy for α-Mannosidosis - Judith Blanz 9:35-9:55 α-Mannosidosis in the Guinea Pig – Pathophysiological, behavioral, and treatment issues - John Hopwood 10:00-10:15 Break α-Mannosidosis (continued) 10:15-10:35 Strategies for translating gene therapy in the brain from rodents to clinical use - John Wolfe 10:35-10:55 Quantitative Imaging of gray matter disease associated with feline α- mannosidosis – Charles Vite β-Mannosidosis 10:55-11:15 β-Mannosidosis mice – A model for the human disease? – Karen Friderici Fucosidosis 11:15-11:30 Intrathecal enzyme infusion therapy in canine fucosidosis – Gauthami Kondagari 11:30-12:00 General Discussion 12:00-1:30 Lunch Afternoon Session, July 26 1:30-2:15 Special Lecture: The Glycoproteinoses: Limited Progress in Molecular Insight still Surpasses Present-day Treatment Efficacy – Jules Leroy Session 2: Animal Models, Pathogenesis, Experimental Therapies (Chair, John Wolfe) -

4Th Glycoproteinoses International Conference Advances in Pathogenesis and Therapy
Program & Abstracts 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ADVANCES IN PATHOGENESIS AND THERAPY ISMRD ST. LOUIS, MISSOURI, UNITED STATES Program & Abstracts I SM R D ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD would like to say A Very Special Thank You to the following organizations and companies who have very generously given donations and sponsorship to support the 4th International Conference on Glycoproteinoses THE PRENILLE EDWARD MALLINCKRODT FOUNDATION JR FOUNDATION MARK HASKINS I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD is very proud to display 10 featured Expression of Hope artworks to be Auctioned at the Gala Dinner. These beautiful prints are from Genzyme’s featured Artwork selection. Contents Welcome 1 SCIENTIFIC COMMITTEE: Stuart Kornfeld ISMRD Mission & Governance 3 (Chair, Scientifi c Planning Committee) Steve Walkley Sara Cathey ISMRD General Information 5 Richard Steet Sean Thomas Ackley, Philippines Miriam Storchli, Switzerland Alessandra d’Azzo ‘Hope’ by Sarah Noble, New Zealand Scientifi c Program 9 FAMILY CONFERENCE COMMITTEE: Family Program for Mucolipidosis 11 Jenny Noble (Conference Organiser) Jackie James (Conference Organiser Family Program For Alpha Mannosidosis /Sialidosis/ 13 - St. Louis) Fucosidosis/Aspartylglucosaminuria Mark Stark John Forman ‘All around the world’ by Zih Yun Li , Taiwan Childrens Program 16 Susan Kester Carolyn Paisley-Dew Tish Adkins Abstracts 17 Sara DeAngelis, Russia Gayle Rose, United States Speaker Profi les 60 Delegates 81 Helen Walker, Australia Nicklas Harkins, Canada Naomi Arai, Japan David Wentworth, Serbia I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts On behalf of the Scientifi c Planning Committee, I want to extend a warm welcome to all the investigators and Welcome! families who have traveled to St. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

A Case of Type I Sialidosis with Osteonecrosis Revealing a New
Original Article Journal of Inborn Errors of Metabolism &Screening 1–3 A Case of Type I Sialidosis With ª The Author(s) 2014 Reprints and permission: Osteonecrosis Revealing a New sagepub.com/journalsPermissions.nav DOI: 10.1177/2326409814543468 Mutation in NEU1 iem.sagepub.com Geoffrey Urbanski1,2, Soumeya Bekri3, Magalie Barth2,4, Christophe Verny4, and Christian Lavigne1,2 Abstract Sialidosis is a rare lysosomal storage disease. The 2 forms described are as follows: the early-onset form, or type II, presents with dysostosis multiplex, while the late-onset form, or type I, does not involve bone in the literature. We report the case of a 42-year- old woman with type I sialidosis who presents with osteonecrosis of both humeral and femoral heads. Molecular study reveals a never listed mutation of NEU1 in exon 5, p.Gly273Asp (c.818G>A), and a second known missense mutation. Keywords sialidosis, bone involvement, NEU1 Introduction developed, at the age of 18 years, a rapidly progressive severe bilateral visual defect leading to blindness. At that time, the Sialidosis (Online Mendelian Inheritance in Man [OMIM] ophthalmologic examination revealed bilateral cherry red spot 256550) is a rare lysosomal storage disease,1 with an estimated in the macula, evolving to macular and optic atrophy associated incidence of 1 in 4 200 000 live births, and it belongs to the group with bilateral cataract. At the age of 32 years, she developed of oligosaccharidoses. Sialidosis is caused by to the recessively myoclonus and epilepsy as grand mal seizure. Myoclonus inherited deficiency of N-acetyl-a-neuraminidase, an acid affected all 4 limbs, but prevailed in the upper limbs, and hydrolase expressed from the gene NEU1, which is located in increased with menstrual cycle and anxiety. -
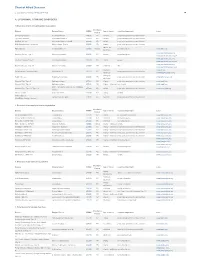
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none -
Lysosomal Disease Network Consortiapedia.Fastercures.Org
Lysosomal Disease Network consortiapedia.fastercures.org Lysosomal Disease Network consortiapedia.fastercures.org/consortia/lysosomal-disease-network/ Research Areas At a Glance Status: Active Consortium Tool Development Year Launched: 2004 Resource Initiating Organization: NIH Rare Disease Basic Research Clinical Research Network Initiator Type: Government Rare disease Location: North America Abstract The combined and integrated efforts of the Lysosomal Disease Network will focus limited resources toward creating a network of centers with expertise in one or more of these diseases in order to solve major challenges in diagnosis, disease management, and therapy. Solutions to these problems will have direct impact on patients suffering from lysosomal diseases and important implications for medical practice. Mission The network is studying the following diseases: alpha-Mannosidosis types I / II Aspartylglucosaminuria Batten disease Batten disease, late infantile beta-Mannosidosis Cystinosis Lysosomal Disease Network - consortiapedia.fastercures.org Page 1/5 Lysosomal Disease Network consortiapedia.fastercures.org Danon disease Fabry disease Farber disease Fucosidosis Galactosialidosis types I / II Gaucher disease GM1-Gangliosidosis types I/II/III GM2-Gangliosidosis Hunter syndrome Hurler syndrome I-cell disease Krabbe disease Maroteaux-Lamy syndrome Metachromatic leukodystrophy Morquio syndrome Mucolipidosis type IV Mucopolysaccharidosis type IX Multiple sulfatase deficiency Niemann-Pick disease Northern Epilepsy Pompe disease pseudo-Hurler -

Allogeneic Hematopoietic SCT for Alpha-Mannosidosis: an Analysis of 17 Patients
Bone Marrow Transplantation (2012) 47, 352–359 & 2012 Macmillan Publishers Limited All rights reserved 0268-3369/12 www.nature.com/bmt ORIGINAL ARTICLE Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients M Mynarek1, J Tolar2, MH Albert3, ML Escolar4, JJ Boelens5, MJ Cowan6, N Finnegan7, A Glomstein8, DA Jacobsohn9,JSKu¨ hl10, H Yabe11, J Kurtzberg12, D Malm13, PJ Orchard2, C Klein1,TLu¨ cke14 and K-W Sykora1 1Hannover Medical School, Department of Pediatric Hematology and Oncology, Hannover, Germany; 2University of Minnesota, Department of Pediatrics, Division of Hematology, Oncology, Blood and Marrow Transplantation, Minneapolis, MN, USA; 3Ludwig Maximilians University, Department of Pediatric Hematology and Oncology, Munich, Germany; 4University of North Carolina at Chapel Hill, Program for Neurodevelopmental Function in Rare Disorders, Chapel Hill, NC, USA; 5University Medical Center Utrecht, Department of Pediatrics: Stem Cell Transplantation Unit, Utrecht, The Netherlands; 6University of California San Francisco, Children’s Hospital, Blood and Marrow Transplant Division, San Francisco, CA, USA; 7Great Ormond Street Hospital for Children NHS Trust, Metabolic Office, London, UK; 8University Hospital Oslo, Department of Pediatrics, Oslo, Norway; 9Children’s National Medical Center, Department for Pediatric Bone Marrow Transplantation, Washington, DC, USA; 10Charite´ University Medicine Berlin, Department of Pediatric Hematology, Oncology and BMT, Berlin, Germany; 11Tokai University School of Medicine, Department of Cell Transplantation and Regenerative Medicine, Shimokasuya, Isehara, Japan; 12Duke University Medical Center, The Pediatric Blood and Marrow Transplant Program, Durham, NC, USA; 13University Hospital of North Norway, Department of Gastroenterology, Tromsoe, Norway and 14Ruhr University Bochum, Department of Neuropediatrics, University Children’s Hospital, Bochum, Germany Alpha-mannosidosis is a rare lysosomal storage disease. -

Human Xc-N-Acetylgalactosaminidase Qx-NAGA)
4585 JMed Genet 1996;33:458-464 Human xc-N-acetylgalactosaminidase Qx-NAGA) deficiency: new mutations and the paradox J Med Genet: first published as 10.1136/jmg.33.6.458 on 1 June 1996. Downloaded from between genotype and phenotype J L M Keulemans, A J J Reuser, M A Kroos, R Willemsen, M M P Hermans, A M W van den Ouweland, J G N de Jong, R A Wevers, W 0 Renier, D Schindler, M J Coll, A Chabas, H Sakuraba, Y Suzuki, 0 P van Diggelen Abstract reported the second independent case of a- Up to now eight patients with a-NAGA NAGA deficiency with an entirely different deficiency have been described. This in- clinical phenotype. This patient had a late onset cludes the newly identified patient re- disease with slight facial coarseness, dis- ported here who died unexpectedly aged seminated angiokeratoma, and mild intellectual II years of hypoxia during convulsions; impairment (IQ= 70), but without neuro- necropsy was not performed. logical symptoms. Unlike the infantile cases, Three patients have been genotyped pre- this patient had prominent vacuolisation in all viously and here we report the mutations dermal cells, most prominently in vascular and in the other five patients, including two lymphatic endothelial cells and eccrine sweat new mutations (S160C and E193X). The gland cells, but also in dermal neural cells and newly identified patient is consanguineous fibroblasts.' The glomerular endothelial cells with the first patients reported with a- but not the epithelial kidney cells are involved NAGA deficiency and neuroaxonal dys- and also blood lymphocytes are vacuolised.6 trophy and they all had the a-NAGA geno- These three patients shared, however, the type E325KIE325K. -

The Myriad Foresight® Carrier Screen
The Myriad Foresight® Carrier Screen 180 Kimball Way | South San Francisco, CA 94080 www.myriadwomenshealth.com | [email protected] | (888) 268-6795 The Myriad Foresight® Carrier Screen - Disease Reference Book 11-beta-hydroxylase-deficient Congenital Adrenal Hyperplasia ...............................................................................................................................................................................8 6-pyruvoyl-tetrahydropterin Synthase Deficiency....................................................................................................................................................................................................10 ABCC8-related Familial Hyperinsulinism..................................................................................................................................................................................................................12 Adenosine Deaminase Deficiency ............................................................................................................................................................................................................................14 Alpha Thalassemia ....................................................................................................................................................................................................................................................16 Alpha-mannosidosis ..................................................................................................................................................................................................................................................18 -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

Lysosomal Storage Disorders: Urine Screening
2460 Mountain Industrial Boulevard | Tucker, Georgia 30084 Phone: 470-378-2200 or 855-831-7447 | Fax: 470-378-2250 eglgenetics.com Lysosomal Storage Disorders: Urine Screening Test Code: BLSDS Turnaround time: 2 weeks (GAGs performed Fri 10 am / Oligos performed Fri 3 pm) CPT Codes: 82542 x1, 82570 x1, 83864 x1, 84275 x1, 84375 x1, 84377 x1 Condition Description Notice: This test has been discontinued EGL no longer accepts samples for this test. For questions, please call: 470-378-2200 In glycoprotein storage diseases (GSDs), certain subtypes of congenital disorders of glycosylation (CDGs), and in the mucolipidoses, there is an accumulation of oligosaccharides, free glycans, glycoamino acids, glycolipid and glycopeptide in the urine. Glycoprotein storage diseases are genetic conditions caused by the body's inability to produce specific enzymes. Normally, the body uses enzymes to process, break down and recycle materials in cells. In individuals with GSD and related diseases, the missing or insufficient enzyme prevents the proper processing and recycling process. This results in the storage of materials, called oligosaccharides or free glycans and glycoamino acids in virtually every cell of the body. As a result, cells do not perform properly and may cause progressive damage throughout the body, including the heart, bones, joints, respiratory system, immune system and central nervous system. While the disease may or may not be apparent at birth, signs and symptoms develop with age as more cells become damaged by the accumulation of materials. The symptoms of these diseases may vary based on syndrome type, and in some cases may resemble a mucopolysaccharidosis. This urinary oligosaccharide and glycan screening uses mass spectrometry (MS), which provides a better sensitivity and specificity than traditional TLC methods.