File Download
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lysosomal Diseases
THE GLYCOPROTEINOSES: SECOND INTERNATIONAL WORKSHOP ON ADVANCES IN PATHOGENESIS AND THERAPY JULY 26-27, 2007 ANN ARBOR, MICHIGAN Scientific Conference Day 1 Morning Sessions, July 26 7:30-8:15 Continental Breakfast 8:15-8:30 Welcome and Introductions, Opening Remarks – Goals of the conference, progress since the last conference - Steven Walkley (with additional comments by officials from ISMRD, NINDS, ORD) 8:30-9:15 Plenary Address Cross-Correction: Looking Back, Looking Forward – Elizabeth Neufeld Session 1: Animal Models, Pathogenesis, Experimental Therapies (Chair, Mark Haskins) α-Mannosidosis 9:15-9:35 Enzyme replacement therapy for α-Mannosidosis - Judith Blanz 9:35-9:55 α-Mannosidosis in the Guinea Pig – Pathophysiological, behavioral, and treatment issues - John Hopwood 10:00-10:15 Break α-Mannosidosis (continued) 10:15-10:35 Strategies for translating gene therapy in the brain from rodents to clinical use - John Wolfe 10:35-10:55 Quantitative Imaging of gray matter disease associated with feline α- mannosidosis – Charles Vite β-Mannosidosis 10:55-11:15 β-Mannosidosis mice – A model for the human disease? – Karen Friderici Fucosidosis 11:15-11:30 Intrathecal enzyme infusion therapy in canine fucosidosis – Gauthami Kondagari 11:30-12:00 General Discussion 12:00-1:30 Lunch Afternoon Session, July 26 1:30-2:15 Special Lecture: The Glycoproteinoses: Limited Progress in Molecular Insight still Surpasses Present-day Treatment Efficacy – Jules Leroy Session 2: Animal Models, Pathogenesis, Experimental Therapies (Chair, John Wolfe) -

4Th Glycoproteinoses International Conference Advances in Pathogenesis and Therapy
Program & Abstracts 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ADVANCES IN PATHOGENESIS AND THERAPY ISMRD ST. LOUIS, MISSOURI, UNITED STATES Program & Abstracts I SM R D ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD would like to say A Very Special Thank You to the following organizations and companies who have very generously given donations and sponsorship to support the 4th International Conference on Glycoproteinoses THE PRENILLE EDWARD MALLINCKRODT FOUNDATION JR FOUNDATION MARK HASKINS I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD is very proud to display 10 featured Expression of Hope artworks to be Auctioned at the Gala Dinner. These beautiful prints are from Genzyme’s featured Artwork selection. Contents Welcome 1 SCIENTIFIC COMMITTEE: Stuart Kornfeld ISMRD Mission & Governance 3 (Chair, Scientifi c Planning Committee) Steve Walkley Sara Cathey ISMRD General Information 5 Richard Steet Sean Thomas Ackley, Philippines Miriam Storchli, Switzerland Alessandra d’Azzo ‘Hope’ by Sarah Noble, New Zealand Scientifi c Program 9 FAMILY CONFERENCE COMMITTEE: Family Program for Mucolipidosis 11 Jenny Noble (Conference Organiser) Jackie James (Conference Organiser Family Program For Alpha Mannosidosis /Sialidosis/ 13 - St. Louis) Fucosidosis/Aspartylglucosaminuria Mark Stark John Forman ‘All around the world’ by Zih Yun Li , Taiwan Childrens Program 16 Susan Kester Carolyn Paisley-Dew Tish Adkins Abstracts 17 Sara DeAngelis, Russia Gayle Rose, United States Speaker Profi les 60 Delegates 81 Helen Walker, Australia Nicklas Harkins, Canada Naomi Arai, Japan David Wentworth, Serbia I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts On behalf of the Scientifi c Planning Committee, I want to extend a warm welcome to all the investigators and Welcome! families who have traveled to St. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

A Case of Type I Sialidosis with Osteonecrosis Revealing a New
Original Article Journal of Inborn Errors of Metabolism &Screening 1–3 A Case of Type I Sialidosis With ª The Author(s) 2014 Reprints and permission: Osteonecrosis Revealing a New sagepub.com/journalsPermissions.nav DOI: 10.1177/2326409814543468 Mutation in NEU1 iem.sagepub.com Geoffrey Urbanski1,2, Soumeya Bekri3, Magalie Barth2,4, Christophe Verny4, and Christian Lavigne1,2 Abstract Sialidosis is a rare lysosomal storage disease. The 2 forms described are as follows: the early-onset form, or type II, presents with dysostosis multiplex, while the late-onset form, or type I, does not involve bone in the literature. We report the case of a 42-year- old woman with type I sialidosis who presents with osteonecrosis of both humeral and femoral heads. Molecular study reveals a never listed mutation of NEU1 in exon 5, p.Gly273Asp (c.818G>A), and a second known missense mutation. Keywords sialidosis, bone involvement, NEU1 Introduction developed, at the age of 18 years, a rapidly progressive severe bilateral visual defect leading to blindness. At that time, the Sialidosis (Online Mendelian Inheritance in Man [OMIM] ophthalmologic examination revealed bilateral cherry red spot 256550) is a rare lysosomal storage disease,1 with an estimated in the macula, evolving to macular and optic atrophy associated incidence of 1 in 4 200 000 live births, and it belongs to the group with bilateral cataract. At the age of 32 years, she developed of oligosaccharidoses. Sialidosis is caused by to the recessively myoclonus and epilepsy as grand mal seizure. Myoclonus inherited deficiency of N-acetyl-a-neuraminidase, an acid affected all 4 limbs, but prevailed in the upper limbs, and hydrolase expressed from the gene NEU1, which is located in increased with menstrual cycle and anxiety. -
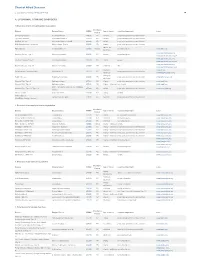
Pdf NTSAD Chart of Allied Diseases
Chart of Allied Diseases Last Updated: Monday, 19 May 2014 17:02 A. LYSOSOMAL STORAGE DISORDERS 1) Disorders of lipid and sphingolipid degradation Inheritance Disease Enzyme Defect OMIM# Age of Onset Cognitive Impairment Links Pattern GM1 Gangliosidosis b-Galactosidase-1 230500 AR variable progressive psychomotor deterioration Tay-Sachs Disease b-Hexosaminidase A 272800 AR variable progressive psychomotor deterioration Sandhoff Disease b-Hexosaminidases A and B 268800 AR variable progressive psychomotor deterioration GM2 Gangliodisosis, AB variant GM2 Activator Protein 272750 AR infancy progressive psychomotor deterioration adolesence - Fabry Disease 8-Galactosidase A 301500 X-linked normal intelligence www.fabry.org adulthood www.gaucherdisease.org, Gaucher Disease, Type 1 Glucocerebrosidase 230800 AR variable normal intelligence www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type II Glucocerebrosidase 230900 AR infancy severe www.gaucherdisease.org.uk www.gaucherdisease.org, Gaucher Disease, Type III Glucocerebrosidase 231000 AR childhood mild www.gaucherdisease.org.uk infancy to www.ulf.org, Metachromatic Leukodystrophy Arylsulfatase A 250100 AR progressive psychomotor deterioration adulthood www.MLDFoundation.org infancy to Krabbe Disease Galactosylceramidase 245200 AR progressive psychomotor deterioration www.huntershope.org adulthood Niemann-Pick, Type A Sphingomyelinase 257200 AR infancy progressive psychomotor deterioration www.nnpdf.org Niemann-Pick, Type B Sphingomyelinase 607616 AR infancy - childhood none -

Allogeneic Hematopoietic SCT for Alpha-Mannosidosis: an Analysis of 17 Patients
Bone Marrow Transplantation (2012) 47, 352–359 & 2012 Macmillan Publishers Limited All rights reserved 0268-3369/12 www.nature.com/bmt ORIGINAL ARTICLE Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients M Mynarek1, J Tolar2, MH Albert3, ML Escolar4, JJ Boelens5, MJ Cowan6, N Finnegan7, A Glomstein8, DA Jacobsohn9,JSKu¨ hl10, H Yabe11, J Kurtzberg12, D Malm13, PJ Orchard2, C Klein1,TLu¨ cke14 and K-W Sykora1 1Hannover Medical School, Department of Pediatric Hematology and Oncology, Hannover, Germany; 2University of Minnesota, Department of Pediatrics, Division of Hematology, Oncology, Blood and Marrow Transplantation, Minneapolis, MN, USA; 3Ludwig Maximilians University, Department of Pediatric Hematology and Oncology, Munich, Germany; 4University of North Carolina at Chapel Hill, Program for Neurodevelopmental Function in Rare Disorders, Chapel Hill, NC, USA; 5University Medical Center Utrecht, Department of Pediatrics: Stem Cell Transplantation Unit, Utrecht, The Netherlands; 6University of California San Francisco, Children’s Hospital, Blood and Marrow Transplant Division, San Francisco, CA, USA; 7Great Ormond Street Hospital for Children NHS Trust, Metabolic Office, London, UK; 8University Hospital Oslo, Department of Pediatrics, Oslo, Norway; 9Children’s National Medical Center, Department for Pediatric Bone Marrow Transplantation, Washington, DC, USA; 10Charite´ University Medicine Berlin, Department of Pediatric Hematology, Oncology and BMT, Berlin, Germany; 11Tokai University School of Medicine, Department of Cell Transplantation and Regenerative Medicine, Shimokasuya, Isehara, Japan; 12Duke University Medical Center, The Pediatric Blood and Marrow Transplant Program, Durham, NC, USA; 13University Hospital of North Norway, Department of Gastroenterology, Tromsoe, Norway and 14Ruhr University Bochum, Department of Neuropediatrics, University Children’s Hospital, Bochum, Germany Alpha-mannosidosis is a rare lysosomal storage disease. -

The Myriad Foresight® Carrier Screen
The Myriad Foresight® Carrier Screen 180 Kimball Way | South San Francisco, CA 94080 www.myriadwomenshealth.com | [email protected] | (888) 268-6795 The Myriad Foresight® Carrier Screen - Disease Reference Book 11-beta-hydroxylase-deficient Congenital Adrenal Hyperplasia ...............................................................................................................................................................................8 6-pyruvoyl-tetrahydropterin Synthase Deficiency....................................................................................................................................................................................................10 ABCC8-related Familial Hyperinsulinism..................................................................................................................................................................................................................12 Adenosine Deaminase Deficiency ............................................................................................................................................................................................................................14 Alpha Thalassemia ....................................................................................................................................................................................................................................................16 Alpha-mannosidosis ..................................................................................................................................................................................................................................................18 -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

Novel Missense Mutations in the Human Lysosomal Sialidase Gene in Sialidosis Patients and Prediction of Structural Alterations of Mutant Enzymes
B.J Hum Jochimsen Genet et(2002) al.: Stetteria 47:29–37 hydrogenophila © Jpn Soc Hum Genet and Springer-Verlag4600/29 2002 ORIGINAL ARTICLE Kohji Itoh · Yasunori Naganawa · Fumiko Matsuzawa Seiichi Aikawa · Hirofumi Doi · Naokazu Sasagasako Takeshi Yamada · Jun-ichi Kira · Takuro Kobayashi Alexey V. Pshezhetsky · Hitoshi Sakuraba Novel missense mutations in the human lysosomal sialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes Received: Stptember 21, 2001 / Accepted: November 2, 2001 Abstract Three novel missense mutations in the human changes including the active site residues responsible for lysosomal sialidase gene causing amino acid substitutions binding the sialic acid carboxylate group. The W240R sub- (P80L, W240R, and P316S) in the coding region were stitution was deduced to influence the molecular surface identified in two Japanese sialidosis patients. One patient structure of a limited region of the constructed models, with a severe, congenital form of type 2 sialidosis was a which was also influenced by previously identified V217M compound heterozygote for 239C-to-T (P80L) and 718T-to- and G243R transversions. C (W240R). The other patient with a mild juvenile-onset phenotype (type 1) was a homozygote for the base substitu- Key words Lysosomal sialidase · Sialidosis · Molecular tion of 946C-to-T (P316S). None of these mutant cDNA modeling · Protective protein/cathepsin A · Galacto- products showed enzymatic activity toward an artificial sialidosis substrate when coexpressed in galactosialidosis fibroblastic cells together with protective protein/cathepsin A (PPCA). All mutants showed a reticular immunofluorescence distri- bution when coexpressed with the PPCA gene in COS-1 Introduction cells, suggesting that the gene products were retained in the endoplasmic reticulum/Golgi area or rapidly degraded Lysosomal sialidase (neuraminidase, EC 3.2.1.18) catalyzes in the lysosomes. -

High Proportion of Mannosidosis and Fucosidosis Among Lysosomal Storage Diseases in Cuba
High proportion of mannosidosis and fucosidosis among lysosomal storage diseases in Cuba C. Menéndez-Sainz1, A. González-Quevedo1, S. González-García1, M. Peña-Sánchez1 and R. Giugliani2 1Neurobiology Department, Institute of Neurology and Neurosurgery, Havana, Cuba 2Serviço de Genética Médica, Hospital das Clínicas de Porto Alegre, Departamento de Genética, Instituto Nacional de Genética Médica Populacional, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brasil Corresponding author: C. Menéndez-Sainz E-mail: [email protected] Genet. Mol. Res. 11 (3): 2352-2359 (2012) Received May 17, 2012 Accepted July 3, 2012 Published August 13, 2012 DOI http://dx.doi.org/10.4238/2012.August.13.9 ABSTRACT. Although lysosomal storage disorders (LSDs) are considered individually rare, as a group they present a non-negligible frequency. Few studies have been made of populational occurrence of LSDs; they have been conducted predominantly on Caucasian populations. We studied the occurrence of LSDs in Cuba. Data from individuals who had been referred to the Institute of Neurology and Neurosurgery in Havana from hospitals all over the country between January 1990 and December 2005 were analyzed. This institute was the only laboratory to provide enzyme-based diagnostic testing for 19 LSDs in Cuba during this period. Occurrence rates were calculated by dividing the number of postnatal diagnoses by the number of births during the study period. The combined occurrence of LSDs in Cuba was 5.6 per 100,000, lower than that reported in other studies conducted on Caucasian populations. The most frequent individual LSDs were: mucopolysaccharidosis type I (1.01 per 100,000) and, Genetics and Molecular Research 11 (3): 2352-2359 (2012) ©FUNPEC-RP www.funpecrp.com.br Lysosomal storage diseases in Cuba 2353 surprisingly, alpha-mannosidosis (0.72 per 100,000) and fucosidosis (0.62 per 100,000). -

Emerging Trends in Transplantation of Inherited Metabolic Diseases
Bone Marrow Transplantation (2008) 41, 99–108 & 2008 Nature Publishing Group All rights reserved 0268-3369/08 $30.00 www.nature.com/bmt REVIEW Emerging trends in transplantation of inherited metabolic diseases VK Prasad and J Kurtzberg The Pediatric Blood and Marrow Transplant Program, Duke University Medical Center, Durham, NC, USA Allogeneic hematopoietic stem cell transplantation received unrelated donor (URD) umbilical cord blood (HSCT) can prolong life and improve its quality in transplants (UCBT).11,12,14–17 patients with inherited metabolic diseases. HSCT offers a In the short term, HSCT has favorably impacted the permanent source of enzyme replacement therapy and clinical outcomes of patients with IMD. However, because also might mediate nonhematopoietic cell regeneration or of limited follow-up, the long-term effects of treatment on repair. Unrelated cord blood is an exciting newer graft the natural history of these disorders and the impact of source for treatment of patients with these fatal disorders, myeloablative therapy are unknown. Recent reports, providing increased access to donors and significant particularly fromUCBT, suggest that there will be clinical efficacy, particularly when transplantation is tremendous benefit if the transplants are performed early performed in early stages. Pre-transplant performance in the course of these diseases, prior to the development of status is highly predictive of overall survival. neurologic and other deficits. Specific host and donor Bone Marrow Transplantation (2008) 41, 99–108; characteristics have impact on immediate transplant out- doi:10.1038/sj.bmt.1705970; published online 7 January 2008 comes (for example, engraftment, GVHD and survival) but Keywords: inherited metabolic diseases; lysosomal and the final measures of success must be benchmarked against peroxisomal storage disease; hematopoietic stem cell clinical development, neurocognitive abilities and non- transplantation; umbilical cord blood transplantation hematopoietic organ function. -
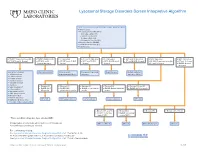
Lysosomal Storage Disorders Screen Interpretive Algorithm
Lysosomal Storage Disorders Screen Interpretive Algorithm LSDS / Lysosomal Storage Disorders Screen, Random, Urine Testing includes: ■ Mucopolysaccharides (MPS): – Dermatan sulfate (DS) – Heparan sulfate (HS) – Keratan sulfate (KS) – Chondroitin 6-sulfate (CS) ■ Oligosaccharides (OLIGO) ■ Ceramide trihexosides (CT) ■ Sulfatides (S) ■ OLIGO: Characteristic profile ■ OLIGO: MLII/III profile ■ S: Abnormal ■ CT and S: Abnormal ■ CT: Abnormal ■ MPS and S: Abnormal ■ MPS: Abnormal ■ MPS: Abnormal ■ CT, MPS and S: Normal ■ CT, MPS and S: ■ CT, MPS and OLIGO: ■ MPS and OLIGO: ■ MPS, OLIGO and S: ■ CT and OLIGO: Normal ■ OLIGO: Characteristic profile ■ CT and S: Normal Normal/abnormal Normal Normal Normal ■ CT and S: Normal ■ OLIGO: Normal/ abnormal One of the following: Mucolipidosis II/III Metachromatic Prosaposin/SaposinB Fabry Disease Multiple sulfatase ■ α-Mannosidosis leukodystrophy (MLD) deficiency deficiency (MSD) ■ β-Mannosidosis ■ Pompe disease ■ Sandhoff disease ■ Schindler disease ■ Sialidosis ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS and CS ■ Galactosialidosis* ■ OLIGO: MPS ■ OLIGO: GM1 ■ OLIGO: α-Fucosidosis ■ OLIGO: Galactosialidosis ■ OLIGO: MPS IVA profile ■ α-Fucosidosis* IVB profile gangliosidosis profile profile profile ■ Mucolipidosis II/III* ■ GM1 gangliosidosis* ■ Morquio A & B* ■ NGYL1 deficiency MPS IVB GM1 gangliosidosis -Fucosidosis Galactosialidosis MPS IVA ■ MOGS-CDG (Congenital α Disorder of Glycosylation-IIb) ■ Elevated DS and HS ■ Elevated DS ■ Elevated HS ■ Elevated DS, HS, CS ■ OLIGO: