Fucosidosis: Ultrastructural Study of Conjunctiva and Skin and Enzyme Analysis of Tears
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mucopolysaccharidoses and Mucolipidoses
J Clin Pathol: first published as 10.1136/jcp.s3-8.1.64 on 1 January 1974. Downloaded from J. clin. Path., 27, Suppl. (Roy. Coll. Path.), 8, 64-93 Mucopolysaccharidoses and mucolipidoses F. VAN HOOF From the Laboratoire de Chimie Physiologique, Universite Catholique de Louvain, and International Institute of Cellular and Molecular Pathology, Brussels, Belgium Many different syndromes are classified as muco- THE CHEMICAL ERA polysaccharidoses, and, despite remarkable progress Chemical studies, performed mainly by groups in the biochemical understanding of these diseases, working with A. Dorfman, in Chicago and K. much remains to be learned and many cases still Meyer, in New York, have provided most of the escape classification. new knowledge in the field by analysis of tissue and Mucopolysaccharidoses are inborn storage dis- urinary mucopolysaccharides in patients (Dorfman eases, characterized by a complex accumulation of and Lorincz, 1957; Meyer, Grumbach, Linker, and mucopolysaccharides and of glycolipids within the Hoffman, 1958; Meyer, Hoffman, Linker, lysosomes. Sixteen human diseases correspond to Grumbach, and Sampson, 1959). These provided the this definition, of which nine have been presently basis for the subdivision of the 'Hurler syndrome' explained by the deficiency of an acid hydrolase. into six subgroups (McKusick, Kaplan, Wise, They are somewhat arbitrarily divided into muco- Hanley, Suddarth, Sevick, and Maumanee, 1965). polysaccharidoses and mucolipidoses. In muco- The possibility that mucopolysaccharidoses could polysaccharidoses, mucopolysaccharides are the result from an excessive biosynthesis of muco- main storage substances, although an abnormal polysaccharides was suggested by Matalon and accumulation of complex lipids is practically always Dorfman (1966). copyright. disclosed at least by the ultiastructural examination. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

Cherry-Red Spot Myoclonus Syndrome and A-Neuraminidase Deficiency
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.43.10.934 on 1 October 1980. Downloaded from Journal of Neurology, Neurosurgery, and Psychiatry, 1980, 43, 934-940 Cherry-red spot myoclonus syndrome and a-neuraminidase deficiency: neurophysiological, pharmacological and biochemical study in an adult S FRANCESCHETTf, G UZIEL, S DI DONATO, L CAIM[*, AND G AVANZfNI From the Departments of Neurophysiology and Neurometabolic Diseases Istituto Neurologico C Besta, Milan, Italy SUMMARY A 22 year old patient with non-familial progressive myoclonus, macular cherry-red spot, moderate cerebellar syndrome and normal intelligence is described. The mycolonus began at the age of 18 years. Focal myoclonus could easily be elicited by voluntary and passive movements, and by touch and electrical stimulation of median nerve. Somatosensory evoked potentials showed a high voltage early component. Jerk-locked averaging of the EEG preceding action myoclonus detected an otherwise hidden, time-related, EEG spike. The myoclonus responded partially but clearly to L-5 hydroxytryptophan plus carbidopa treatment. Biochemical study showed an a-neuraminidase deficiency in cultured fibroblasts: the decrease in this enzyme activity was compared to that found in a patient affected by mucolipidosis III. Protected by copyright. A slowly progressing neurological syndrome that Case report combines action sensitive and stimulus-sensitive myoclonus with cherry-red spots at the macula The patient was an Italian male aged 21 years. in patients with a-neuraminidase deficiency has There was nothing significant in the family history. In The patient was cyanotic at birth but his psycho- been described recently.'-3 these patients, there motor development 'had been normal. -

The Myriad Foresight® Carrier Screen
The Myriad Foresight® Carrier Screen 180 Kimball Way | South San Francisco, CA 94080 www.myriadwomenshealth.com | [email protected] | (888) 268-6795 The Myriad Foresight® Carrier Screen - Disease Reference Book 11-beta-hydroxylase-deficient Congenital Adrenal Hyperplasia ...............................................................................................................................................................................8 6-pyruvoyl-tetrahydropterin Synthase Deficiency....................................................................................................................................................................................................10 ABCC8-related Familial Hyperinsulinism..................................................................................................................................................................................................................12 Adenosine Deaminase Deficiency ............................................................................................................................................................................................................................14 Alpha Thalassemia ....................................................................................................................................................................................................................................................16 Alpha-mannosidosis ..................................................................................................................................................................................................................................................18 -

I-Cell Disease): a Rare Condition Resembling Hurler Syndrome: a Case Report S Yang, TW Yeung, HY Lau Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong
Hong Kong J Radiol. 2020;23:39-43 | https://doi.org/10.12809/hkjr2017142 CASE REPORT Mucolipidosis Type II (I-cell Disease): A Rare Condition Resembling Hurler Syndrome: A Case Report S Yang, TW Yeung, HY Lau Department of Radiology, Tuen Mun Hospital, Tuen Mun, Hong Kong INTRODUCTION CASE REPORT Mucolipidosis type II (I-cell disease) is a rare autosomal A full term appropriate for gestational age recessive disorder of lysosomal metabolism with consanguineous Pakistani girl was born by emergency progressive multisystem deterioration that leads to Caesarean section for previous Caesarean section and death before or in early childhood. This disease was premature rupture of membrane. Antenatal history was first described in 1967 by Leroy and DeMars.1 Children unremarkable. There was a strong family history of with I-cell disease share many clinical and radiological I-cell disease. In the extended family, eight children had features with Hurler syndrome although there are distinct been diagnosed with the disease and an elder brother differences. Presentation of I-cell disease is earlier with a died from it at age 7 years. The patient presented with shorter clinical course, the radiological changes are more respiratory distress and was diagnosed to have transient profound, and the biochemical features are distinctive. tachypnoea of newborn. Initial chest radiograph The clinical course of I-cell disease is characterised revealed abnormal widened oar-shaped ribs (Figure 1). by progressive failure to thrive and developmental A skeletal survey -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

High Proportion of Mannosidosis and Fucosidosis Among Lysosomal Storage Diseases in Cuba
High proportion of mannosidosis and fucosidosis among lysosomal storage diseases in Cuba C. Menéndez-Sainz1, A. González-Quevedo1, S. González-García1, M. Peña-Sánchez1 and R. Giugliani2 1Neurobiology Department, Institute of Neurology and Neurosurgery, Havana, Cuba 2Serviço de Genética Médica, Hospital das Clínicas de Porto Alegre, Departamento de Genética, Instituto Nacional de Genética Médica Populacional, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brasil Corresponding author: C. Menéndez-Sainz E-mail: [email protected] Genet. Mol. Res. 11 (3): 2352-2359 (2012) Received May 17, 2012 Accepted July 3, 2012 Published August 13, 2012 DOI http://dx.doi.org/10.4238/2012.August.13.9 ABSTRACT. Although lysosomal storage disorders (LSDs) are considered individually rare, as a group they present a non-negligible frequency. Few studies have been made of populational occurrence of LSDs; they have been conducted predominantly on Caucasian populations. We studied the occurrence of LSDs in Cuba. Data from individuals who had been referred to the Institute of Neurology and Neurosurgery in Havana from hospitals all over the country between January 1990 and December 2005 were analyzed. This institute was the only laboratory to provide enzyme-based diagnostic testing for 19 LSDs in Cuba during this period. Occurrence rates were calculated by dividing the number of postnatal diagnoses by the number of births during the study period. The combined occurrence of LSDs in Cuba was 5.6 per 100,000, lower than that reported in other studies conducted on Caucasian populations. The most frequent individual LSDs were: mucopolysaccharidosis type I (1.01 per 100,000) and, Genetics and Molecular Research 11 (3): 2352-2359 (2012) ©FUNPEC-RP www.funpecrp.com.br Lysosomal storage diseases in Cuba 2353 surprisingly, alpha-mannosidosis (0.72 per 100,000) and fucosidosis (0.62 per 100,000). -

A GNPTAB Nonsense Variant Is Associated with Feline Mucolipidosis II (I-Cell Disease) Ping Wang* , Hamutal Mazrier, Jessica Caverly Rae, Karthik Raj and Urs Giger
Wang et al. BMC Veterinary Research (2018) 14:416 https://doi.org/10.1186/s12917-018-1728-1 RESEARCH ARTICLE Open Access A GNPTAB nonsense variant is associated with feline mucolipidosis II (I-cell disease) Ping Wang* , Hamutal Mazrier, Jessica Caverly Rae, Karthik Raj and Urs Giger Abstract Background: Mucolipidosis II (ML II; I-cell disease) is caused by a deficiency of N-acetylglucosamine-1-phosphotransferase (GNPTAB; EC 2.7.8.17), which leads to a failure to internalize acid hydrolases into lysosomes for proper catabolism of various substances. This is an autosomal recessive lysosomal storage disease and causes severe progressive neuropathy and oculoskeletal dysfunction in humans (OMIM 252500). A naturally occurring disease model has been reported in juvenile domestic cats (OMIA 001248–9685) with clinical signs similar to human patients. We investigated the molecular genetic basis of ML II in a colony of affected cats by sequencing the coding and regulatory regions of GNPTAB from affected and clinically healthy related and unrelated domestic cats and compared the sequences to the published feline genome sequence (NCBI-RefSeq accession no. XM_003989173.4, Gene ID: 101100231). Results: All affected cats were homozygous for a single base substitution (c.2644C > T) in exon 13 of GNPTAB. This variant results in a premature stop codon (p.Gln882*) which predicts severe truncation and complete dysfunction of the GNPTAB enzyme. About 140 GNPTAB variants have been described in human ML II patients, with 41.3% nonsense/missense mutations, nine occurring in the same gene region as in this feline model. Restriction fragment length polymorphism and allelic discrimination real-time polymerase chain reaction assays accurately differentiated between clear, asymptomatic carriers and homozygous affected cats. -

Emerging Trends in Transplantation of Inherited Metabolic Diseases
Bone Marrow Transplantation (2008) 41, 99–108 & 2008 Nature Publishing Group All rights reserved 0268-3369/08 $30.00 www.nature.com/bmt REVIEW Emerging trends in transplantation of inherited metabolic diseases VK Prasad and J Kurtzberg The Pediatric Blood and Marrow Transplant Program, Duke University Medical Center, Durham, NC, USA Allogeneic hematopoietic stem cell transplantation received unrelated donor (URD) umbilical cord blood (HSCT) can prolong life and improve its quality in transplants (UCBT).11,12,14–17 patients with inherited metabolic diseases. HSCT offers a In the short term, HSCT has favorably impacted the permanent source of enzyme replacement therapy and clinical outcomes of patients with IMD. However, because also might mediate nonhematopoietic cell regeneration or of limited follow-up, the long-term effects of treatment on repair. Unrelated cord blood is an exciting newer graft the natural history of these disorders and the impact of source for treatment of patients with these fatal disorders, myeloablative therapy are unknown. Recent reports, providing increased access to donors and significant particularly fromUCBT, suggest that there will be clinical efficacy, particularly when transplantation is tremendous benefit if the transplants are performed early performed in early stages. Pre-transplant performance in the course of these diseases, prior to the development of status is highly predictive of overall survival. neurologic and other deficits. Specific host and donor Bone Marrow Transplantation (2008) 41, 99–108; characteristics have impact on immediate transplant out- doi:10.1038/sj.bmt.1705970; published online 7 January 2008 comes (for example, engraftment, GVHD and survival) but Keywords: inherited metabolic diseases; lysosomal and the final measures of success must be benchmarked against peroxisomal storage disease; hematopoietic stem cell clinical development, neurocognitive abilities and non- transplantation; umbilical cord blood transplantation hematopoietic organ function. -
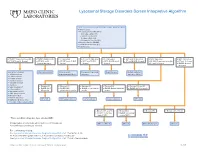
Lysosomal Storage Disorders Screen Interpretive Algorithm
Lysosomal Storage Disorders Screen Interpretive Algorithm LSDS / Lysosomal Storage Disorders Screen, Random, Urine Testing includes: ■ Mucopolysaccharides (MPS): – Dermatan sulfate (DS) – Heparan sulfate (HS) – Keratan sulfate (KS) – Chondroitin 6-sulfate (CS) ■ Oligosaccharides (OLIGO) ■ Ceramide trihexosides (CT) ■ Sulfatides (S) ■ OLIGO: Characteristic profile ■ OLIGO: MLII/III profile ■ S: Abnormal ■ CT and S: Abnormal ■ CT: Abnormal ■ MPS and S: Abnormal ■ MPS: Abnormal ■ MPS: Abnormal ■ CT, MPS and S: Normal ■ CT, MPS and S: ■ CT, MPS and OLIGO: ■ MPS and OLIGO: ■ MPS, OLIGO and S: ■ CT and OLIGO: Normal ■ OLIGO: Characteristic profile ■ CT and S: Normal Normal/abnormal Normal Normal Normal ■ CT and S: Normal ■ OLIGO: Normal/ abnormal One of the following: Mucolipidosis II/III Metachromatic Prosaposin/SaposinB Fabry Disease Multiple sulfatase ■ α-Mannosidosis leukodystrophy (MLD) deficiency deficiency (MSD) ■ β-Mannosidosis ■ Pompe disease ■ Sandhoff disease ■ Schindler disease ■ Sialidosis ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS and CS ■ Galactosialidosis* ■ OLIGO: MPS ■ OLIGO: GM1 ■ OLIGO: α-Fucosidosis ■ OLIGO: Galactosialidosis ■ OLIGO: MPS IVA profile ■ α-Fucosidosis* IVB profile gangliosidosis profile profile profile ■ Mucolipidosis II/III* ■ GM1 gangliosidosis* ■ Morquio A & B* ■ NGYL1 deficiency MPS IVB GM1 gangliosidosis -Fucosidosis Galactosialidosis MPS IVA ■ MOGS-CDG (Congenital α Disorder of Glycosylation-IIb) ■ Elevated DS and HS ■ Elevated DS ■ Elevated HS ■ Elevated DS, HS, CS ■ OLIGO: -

Lysosomal Storage Disease Panel by Next-Generation Sequencing
TEST ID: LSDP LYSOSOMAL STORAGE DISEASE PANEL BY NEXT-GENERATION SEQUENCING USEFUL FOR REFERENCE VALUES } Follow up for abnormal biochemical results and confirmation of suspected lysosomal storage An interpretive report will be disease (LSD) provided. } Identifying mutations within genes known to be associated with lysosomal storage disease, allowing for predictive testing of at-risk family members ANALYTIC TIME 4 weeks CLINICAL INFORMATION Lysosomal storage diseases (LSDs) encompass a group of over 40 inherited biochemical diseases in which genetic mutations cause defective lysosomal functioning. Lysosomes perform catabolic functions for cells, which is accomplished through activity of various proteins such as lysosomal enzymes, transport proteins, and other proteins. Functional deficits in these proteins cause an accumulation of substrates in cells leading to progressive organ dysfunction. This leads to variable clinical features that can affect the cardiovascular, neurological, ocular, and skeletal systems, among others. Clinical features are dependent on the amount and location of the substrate accumulation, but may include the following: characteristic facial features (coarse features), hepatomegaly, deafness, vision loss, abnormal skeletal findings, hydrops fetalis, ataxia, hypotonia, developmental delay/regression, and intellectual disability. Age of onset is variable, with symptoms presenting from the prenatal period to adulthood, but generally LSDs are progressive and cause significant morbidity and mortality with a decreased lifespan. Enzyme replacement therapy and oral substrate inhibitors are therapeutic options for some LSDs. LSDs are inherited in an autosomal recessive manner with the exception of Hunter, Fabry, and Danon diseases, which are X-linked. There are some founder mutations associated with particular LSDs in the Ashkenazi Jewish and Finnish populations, leading to an increased carrier frequency for some. -
DPP-IV) Activity in Plasma from Patients with Various Lysosomal Diseases
diagnostics Article Elevated Dipeptidyl Peptidase IV (DPP-IV) Activity in Plasma from Patients with Various Lysosomal Diseases Agnieszka Ługowska 1,* , Galina Baydakova 2, Alex Ilyushkina 2, Ekaterina Zakharova 2, Hanna Mierzewska 3, Krystyna Szyma ´nska 4 , Jolanta Wierzba 5, Jolanta Kubalska 1, Ałła Graban 6, Tomasz Kmie´c 7, Barbara Perkowska-Sumiła 8, Anna Tylki-Szyma´nska 8 and Małgorzata Bednarska-Makaruk 1 1 Department of Genetics, Institute of Psychiatry and Neurology, 02-957 Warsaw, Poland; [email protected] (J.K.); [email protected] (M.B.-M.) 2 Research Centre for Medical Genetics, Federal State Budgetary Institution, 115478 Moscow, Russia; [email protected] (G.B.); [email protected] (A.I.); [email protected] (E.Z.) 3 Department of Child and Adolescent Neurology, Institute of Mother and Child, 01-211 Warsaw, Poland; [email protected] 4 Mossakowski Medical Research Center, Department of Experimental and Clinical Neuropathology, Polish Academy of Sciences, 02-106 Warsaw, Poland; [email protected] 5 Department of Internal and Pediatric Nursing, Institute of Nursing and Midwifery, Medical University of Gda´nsk,80-210 Gda´nsk,Poland; [email protected] 6 1st Department of Neurology, Institute of Psychiatry and Neurology, 02-957 Warsaw, Poland; [email protected] 7 Department of Neurology and Epileptology, The Children’s Memorial Health Institute, 04-730 Warsaw, Poland; [email protected] 8 Citation: Ługowska, A.; Department of Pediatrics, Nutrition and Metabolic Diseases, The Children’s Memorial Health Institute, Baydakova, G.; Ilyushkina, A.; 04-730 Warsaw, Poland; [email protected] (B.P.-S.); [email protected] (A.T.-S.) Zakharova, E.; Mierzewska, H.; * Correspondence: [email protected]; Tel.: +48-22-4582-610; Fax: +48-22-8589169 Szyma´nska,K.; Wierzba, J.; Kubalska, J.; Graban, A.; Kmie´c,T.; Abstract: Increased activity of dipeptidyl peptidase IV (DPP-IV) was reported earlier in patients et al.