Alpha-Fucosidosis ¬タモ Two Brothers Presenting with Dysostosis Multiplex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neuroradiologic Findings in Fucosidosis, a Rare Lysosomal Storage Disease
Neuroradiologic Findings in Fucosidosis, a Rare Lysosomal Storage Disease James M. Provenzale, Daniel P. Barboriak, and Katherine Sims Summary: Fucosidosis is a rare lysosomal storage disorder with vacuolated secondary lysosomes containing some fine the clinical features of mental retardation, cardiomegaly, dysos- fibrillar material and lamellated membrane structures. tosis multiplex, progressive neurologic deterioration, and early A magnetic resonance (MR) examination at the age of death. The neuroradiologic findings in two patients are reported, 10 years showed confluent regions of hyperintense signal and include abnormalities within the globus pallidus (both pa- on T2-weighted images in the periventricular regions, tients) and periventricular white matter (one patient). most prominent surrounding the atria of the lateral ventri- cles. Hyperintense signal was noted in the globus pallidus Index terms: Brain, metabolism; Degenerative disease, Pediatric on T1-weighted images, with corresponding hypointense neuroradiology signal on T2-weighted images (Fig 1). Fucosidosis is a rare metabolic disorder caused by decreased amounts of the enzyme Case 2 a-L-fucosidase, which results in the accumula- A 2-year-old boy was examined for speech delay and tion of fucose-rich storage products within psychomotor retardation. He was born at term after a many organs, including the brain. Patients with normal pregnancy, and appropriate development oc- this disorder usually have delayed intellectual curred during the first year of life. However, by age 2 years, and motor development, and often die within the patient had not developed speech, exhibited autistic the first decade of life. Computed tomographic behavior, and performed motor tasks poorly. Physical ex- (CT) findings have been reported in a few cases amination findings included coarsened facial features, nar- (1, 2). -

4Th Glycoproteinoses International Conference Advances in Pathogenesis and Therapy
Program & Abstracts 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ADVANCES IN PATHOGENESIS AND THERAPY ISMRD ST. LOUIS, MISSOURI, UNITED STATES Program & Abstracts I SM R D ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD would like to say A Very Special Thank You to the following organizations and companies who have very generously given donations and sponsorship to support the 4th International Conference on Glycoproteinoses THE PRENILLE EDWARD MALLINCKRODT FOUNDATION JR FOUNDATION MARK HASKINS I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts ISMRD is very proud to display 10 featured Expression of Hope artworks to be Auctioned at the Gala Dinner. These beautiful prints are from Genzyme’s featured Artwork selection. Contents Welcome 1 SCIENTIFIC COMMITTEE: Stuart Kornfeld ISMRD Mission & Governance 3 (Chair, Scientifi c Planning Committee) Steve Walkley Sara Cathey ISMRD General Information 5 Richard Steet Sean Thomas Ackley, Philippines Miriam Storchli, Switzerland Alessandra d’Azzo ‘Hope’ by Sarah Noble, New Zealand Scientifi c Program 9 FAMILY CONFERENCE COMMITTEE: Family Program for Mucolipidosis 11 Jenny Noble (Conference Organiser) Jackie James (Conference Organiser Family Program For Alpha Mannosidosis /Sialidosis/ 13 - St. Louis) Fucosidosis/Aspartylglucosaminuria Mark Stark John Forman ‘All around the world’ by Zih Yun Li , Taiwan Childrens Program 16 Susan Kester Carolyn Paisley-Dew Tish Adkins Abstracts 17 Sara DeAngelis, Russia Gayle Rose, United States Speaker Profi les 60 Delegates 81 Helen Walker, Australia Nicklas Harkins, Canada Naomi Arai, Japan David Wentworth, Serbia I SM R D 4TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE 2015 ADVANCES IN PATHOGENESIS AND THERAPY Program & Abstracts On behalf of the Scientifi c Planning Committee, I want to extend a warm welcome to all the investigators and Welcome! families who have traveled to St. -

Mucopolysaccharidoses and Mucolipidoses
J Clin Pathol: first published as 10.1136/jcp.s3-8.1.64 on 1 January 1974. Downloaded from J. clin. Path., 27, Suppl. (Roy. Coll. Path.), 8, 64-93 Mucopolysaccharidoses and mucolipidoses F. VAN HOOF From the Laboratoire de Chimie Physiologique, Universite Catholique de Louvain, and International Institute of Cellular and Molecular Pathology, Brussels, Belgium Many different syndromes are classified as muco- THE CHEMICAL ERA polysaccharidoses, and, despite remarkable progress Chemical studies, performed mainly by groups in the biochemical understanding of these diseases, working with A. Dorfman, in Chicago and K. much remains to be learned and many cases still Meyer, in New York, have provided most of the escape classification. new knowledge in the field by analysis of tissue and Mucopolysaccharidoses are inborn storage dis- urinary mucopolysaccharides in patients (Dorfman eases, characterized by a complex accumulation of and Lorincz, 1957; Meyer, Grumbach, Linker, and mucopolysaccharides and of glycolipids within the Hoffman, 1958; Meyer, Hoffman, Linker, lysosomes. Sixteen human diseases correspond to Grumbach, and Sampson, 1959). These provided the this definition, of which nine have been presently basis for the subdivision of the 'Hurler syndrome' explained by the deficiency of an acid hydrolase. into six subgroups (McKusick, Kaplan, Wise, They are somewhat arbitrarily divided into muco- Hanley, Suddarth, Sevick, and Maumanee, 1965). polysaccharidoses and mucolipidoses. In muco- The possibility that mucopolysaccharidoses could polysaccharidoses, mucopolysaccharides are the result from an excessive biosynthesis of muco- main storage substances, although an abnormal polysaccharides was suggested by Matalon and accumulation of complex lipids is practically always Dorfman (1966). copyright. disclosed at least by the ultiastructural examination. -

International Conference
5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE Rome, Italy November 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE PROGRAM & ABSTRACTS 5TH GLYCOPROTEINOSES INTERNATIONAL CONFERENCE ROME, ITALY NOVEMBER 1-4 2017 EMBRACING INNOVATION ADVANCING THE CURE ISMRD would like to say a very special thank you to the following organizations and companies who have very generously given donations to support the 5th International Conference on Glycoproteinoses. ISMRD is an internationally focused not-for-profi t organization whose mission is to advocate for families and patients aff ected by one of the following disorders. Alpha-Mannosidosis THE WAGNER FOUNDATION Aspartylglucosaminuria Beta-Mannosidosis Fucosidosis Galactosialidosis ISMRD is very grateful for all the help and support that Symposia has given us Sialidosis (Mucolipidosis I) in the organization of our Conference on-the-ground support in Rome. Mucolipidosis II, II/III, III alpha/beta Mucolipidosis III Gamma Schindler Disease EMBRACING INNOVATION ADVANCING THE CURE SCIENTIFIC COMMITTEE: Alessandra d’Azzo CHAIR Contents Amelia Morrone Italy Richard Steet USA Welcome 2 Heather Flanagan-Steet USA ISMRD Mission & Governance 4 Dag Malm Norway ISMRD General Information 6 Thomas Braulke Dedicated to helping patients Germany in the rare disease community Stuart Kornfeld with unmet medical needs Scientifi c Program 10 USA Ultragenyx Pharmaceutical Inc. is a clinical-stage Family Program 14 ISMRD CONFERENCE biopharmaceutical company committed to creating new COMMITTEE: therapeutics to combat serious, -

A Case of Type I Sialidosis with Osteonecrosis Revealing a New
Original Article Journal of Inborn Errors of Metabolism &Screening 1–3 A Case of Type I Sialidosis With ª The Author(s) 2014 Reprints and permission: Osteonecrosis Revealing a New sagepub.com/journalsPermissions.nav DOI: 10.1177/2326409814543468 Mutation in NEU1 iem.sagepub.com Geoffrey Urbanski1,2, Soumeya Bekri3, Magalie Barth2,4, Christophe Verny4, and Christian Lavigne1,2 Abstract Sialidosis is a rare lysosomal storage disease. The 2 forms described are as follows: the early-onset form, or type II, presents with dysostosis multiplex, while the late-onset form, or type I, does not involve bone in the literature. We report the case of a 42-year- old woman with type I sialidosis who presents with osteonecrosis of both humeral and femoral heads. Molecular study reveals a never listed mutation of NEU1 in exon 5, p.Gly273Asp (c.818G>A), and a second known missense mutation. Keywords sialidosis, bone involvement, NEU1 Introduction developed, at the age of 18 years, a rapidly progressive severe bilateral visual defect leading to blindness. At that time, the Sialidosis (Online Mendelian Inheritance in Man [OMIM] ophthalmologic examination revealed bilateral cherry red spot 256550) is a rare lysosomal storage disease,1 with an estimated in the macula, evolving to macular and optic atrophy associated incidence of 1 in 4 200 000 live births, and it belongs to the group with bilateral cataract. At the age of 32 years, she developed of oligosaccharidoses. Sialidosis is caused by to the recessively myoclonus and epilepsy as grand mal seizure. Myoclonus inherited deficiency of N-acetyl-a-neuraminidase, an acid affected all 4 limbs, but prevailed in the upper limbs, and hydrolase expressed from the gene NEU1, which is located in increased with menstrual cycle and anxiety. -

Allo HCT for Metabolic Disorders and Severe Osteopetrosis Status: Recruiting
MT2013-31: Allo HCT for Metabolic Disorders and Severe Osteopetrosis Status: Recruiting Eligibility Criteria Sex: All Age: up to 55 Years old This study is NOT accepting healthy volunteers Inclusion Criteria: • 0 through 55 years of age • Adequate graft available • Adequate organ function • Eligible Diseases: • Mucopolysaccharidosis Disorders: • MPS IH (Hurler syndrome) • MPS II (Hunter syndrome) if the patient has no or minimal evidence of symptomatic neurologic disease but is expected to have a neurologic phenotype • MPS VI (Maroteaux-Lamy syndrome) • MPS VII (Sly syndrome) • Glycoprotein Metabolic Disorders: • Alpha mannosidosis • Fucosidosis • Aspartylglucosaminuria • Sphingolipidoses and Recessive Leukodystrophies: • Globoid cell leukodystrophy • Metachromatic leukodystrophy • Niemann-Pick B patients (sphingomyelin deficiency) • Niemann-Pick C subtype 2 • Peroxisomal Disorders: • Adrenoleukodystrophy with cerebral involvement • Zellweger syndrome • Neonatal Adrenoleukodystrophy • Infantile Refsum disease • Acyl-CoA-Oxidase Deficiency • D-Bifunctional enzyme deficiency • Multifunctional enzyme deficiency • Alpha-methylacyl-CoA Racmase Deficiency (AMACRD) • Mitochondrial Neurogastrointestingal Encephalopathy (MNGIE) • Severe Osteopetrosis (OP) • Hereditary Leukoencephalopathy with axonal spheroids (HDLS; CSF1R mutation) • Other Inherited Metabolic Disorders (IMD): Patients will also be considered who have other life-threatening, rare lysosomal, peroxisomal or other similar inherited disorders characterized by white matter disease or -

SSIEM Classification of Inborn Errors of Metabolism 2011
SSIEM classification of Inborn Errors of Metabolism 2011 Disease group / disease ICD10 OMIM 1. Disorders of amino acid and peptide metabolism 1.1. Urea cycle disorders and inherited hyperammonaemias 1.1.1. Carbamoylphosphate synthetase I deficiency 237300 1.1.2. N-Acetylglutamate synthetase deficiency 237310 1.1.3. Ornithine transcarbamylase deficiency 311250 S Ornithine carbamoyltransferase deficiency 1.1.4. Citrullinaemia type1 215700 S Argininosuccinate synthetase deficiency 1.1.5. Argininosuccinic aciduria 207900 S Argininosuccinate lyase deficiency 1.1.6. Argininaemia 207800 S Arginase I deficiency 1.1.7. HHH syndrome 238970 S Hyperammonaemia-hyperornithinaemia-homocitrullinuria syndrome S Mitochondrial ornithine transporter (ORNT1) deficiency 1.1.8. Citrullinemia Type 2 603859 S Aspartate glutamate carrier deficiency ( SLC25A13) S Citrin deficiency 1.1.9. Hyperinsulinemic hypoglycemia and hyperammonemia caused by 138130 activating mutations in the GLUD1 gene 1.1.10. Other disorders of the urea cycle 238970 1.1.11. Unspecified hyperammonaemia 238970 1.2. Organic acidurias 1.2.1. Glutaric aciduria 1.2.1.1. Glutaric aciduria type I 231670 S Glutaryl-CoA dehydrogenase deficiency 1.2.1.2. Glutaric aciduria type III 231690 1.2.2. Propionic aciduria E711 232000 S Propionyl-CoA-Carboxylase deficiency 1.2.3. Methylmalonic aciduria E711 251000 1.2.3.1. Methylmalonyl-CoA mutase deficiency 1.2.3.2. Methylmalonyl-CoA epimerase deficiency 251120 1.2.3.3. Methylmalonic aciduria, unspecified 1.2.4. Isovaleric aciduria E711 243500 S Isovaleryl-CoA dehydrogenase deficiency 1.2.5. Methylcrotonylglycinuria E744 210200 S Methylcrotonyl-CoA carboxylase deficiency 1.2.6. Methylglutaconic aciduria E712 250950 1.2.6.1. Methylglutaconic aciduria type I E712 250950 S 3-Methylglutaconyl-CoA hydratase deficiency 1.2.6.2. -

Collectively Common, Individually Rare Many
OUTLOOK LYSOSOMAL STORAGE DISORDERS MYRIAD MALADIES u A tophag os om e Mutations affecting an es enzyme that labels other som do 3 enzymes for delivery to n the lysosome result in E 4 mucolipidosis-II Defects in a protein that helps endosomes and autophagosomes fuse with the lysosome cause Danon disease Defects in processing enzymes can cause Ly undigested material to Go soso lgi 2 me accumulate, such as in Gaucher’s disease and 5 Hunter syndrome En doplas mic retic ulum 1 Failure of processing in the endoplasmic reticulum can inac- 6 tivate a whole class Loss of transport proteins of enzymes, such as causes the lysosome to retain molecular building blocks in Nucleus in multiple sulfatase deficiency diseases such as cystinosis or sialic acid storage disease MANY MOVING PARTS The lysosome uses specialized enzymes to help cells to digest external biological materials and recycle defective proteins and damaged cellular machinery. Many LSDs arise from mutations in the genes that encode those enzymes, but there are numerous other ways in which this process can break down (red stars). 1 The endoplasmic The Golgi apparatus Endosomes transport The autophagosome Within the lysosome, Molecular building blocks reticulum performs the further processes and enzymes from the Golgi delivers damaged enzymes convert are released into cell for initial processing of labels specific enzymes and materials from organelles and misfolded molecules such as sugars, reuse newly synthesized for delivery to the outside the cell to the proteins to the lysosome proteins and lipids into lysosomal enzymes lysosome lysosome for recycling simpler building blocks COLLECTIVELY COMMON, INDIVIDUALLY RARE LSDs are not especially rare; estimates suggest that 1 in just over 5,000 newborns newborns worldwide, and the rarest have been described only a handful of times. -

Novel Missense Mutations in the Human Lysosomal Sialidase Gene in Sialidosis Patients and Prediction of Structural Alterations of Mutant Enzymes
B.J Hum Jochimsen Genet et(2002) al.: Stetteria 47:29–37 hydrogenophila © Jpn Soc Hum Genet and Springer-Verlag4600/29 2002 ORIGINAL ARTICLE Kohji Itoh · Yasunori Naganawa · Fumiko Matsuzawa Seiichi Aikawa · Hirofumi Doi · Naokazu Sasagasako Takeshi Yamada · Jun-ichi Kira · Takuro Kobayashi Alexey V. Pshezhetsky · Hitoshi Sakuraba Novel missense mutations in the human lysosomal sialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes Received: Stptember 21, 2001 / Accepted: November 2, 2001 Abstract Three novel missense mutations in the human changes including the active site residues responsible for lysosomal sialidase gene causing amino acid substitutions binding the sialic acid carboxylate group. The W240R sub- (P80L, W240R, and P316S) in the coding region were stitution was deduced to influence the molecular surface identified in two Japanese sialidosis patients. One patient structure of a limited region of the constructed models, with a severe, congenital form of type 2 sialidosis was a which was also influenced by previously identified V217M compound heterozygote for 239C-to-T (P80L) and 718T-to- and G243R transversions. C (W240R). The other patient with a mild juvenile-onset phenotype (type 1) was a homozygote for the base substitu- Key words Lysosomal sialidase · Sialidosis · Molecular tion of 946C-to-T (P316S). None of these mutant cDNA modeling · Protective protein/cathepsin A · Galacto- products showed enzymatic activity toward an artificial sialidosis substrate when coexpressed in galactosialidosis fibroblastic cells together with protective protein/cathepsin A (PPCA). All mutants showed a reticular immunofluorescence distri- bution when coexpressed with the PPCA gene in COS-1 Introduction cells, suggesting that the gene products were retained in the endoplasmic reticulum/Golgi area or rapidly degraded Lysosomal sialidase (neuraminidase, EC 3.2.1.18) catalyzes in the lysosomes. -

High Proportion of Mannosidosis and Fucosidosis Among Lysosomal Storage Diseases in Cuba
High proportion of mannosidosis and fucosidosis among lysosomal storage diseases in Cuba C. Menéndez-Sainz1, A. González-Quevedo1, S. González-García1, M. Peña-Sánchez1 and R. Giugliani2 1Neurobiology Department, Institute of Neurology and Neurosurgery, Havana, Cuba 2Serviço de Genética Médica, Hospital das Clínicas de Porto Alegre, Departamento de Genética, Instituto Nacional de Genética Médica Populacional, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brasil Corresponding author: C. Menéndez-Sainz E-mail: [email protected] Genet. Mol. Res. 11 (3): 2352-2359 (2012) Received May 17, 2012 Accepted July 3, 2012 Published August 13, 2012 DOI http://dx.doi.org/10.4238/2012.August.13.9 ABSTRACT. Although lysosomal storage disorders (LSDs) are considered individually rare, as a group they present a non-negligible frequency. Few studies have been made of populational occurrence of LSDs; they have been conducted predominantly on Caucasian populations. We studied the occurrence of LSDs in Cuba. Data from individuals who had been referred to the Institute of Neurology and Neurosurgery in Havana from hospitals all over the country between January 1990 and December 2005 were analyzed. This institute was the only laboratory to provide enzyme-based diagnostic testing for 19 LSDs in Cuba during this period. Occurrence rates were calculated by dividing the number of postnatal diagnoses by the number of births during the study period. The combined occurrence of LSDs in Cuba was 5.6 per 100,000, lower than that reported in other studies conducted on Caucasian populations. The most frequent individual LSDs were: mucopolysaccharidosis type I (1.01 per 100,000) and, Genetics and Molecular Research 11 (3): 2352-2359 (2012) ©FUNPEC-RP www.funpecrp.com.br Lysosomal storage diseases in Cuba 2353 surprisingly, alpha-mannosidosis (0.72 per 100,000) and fucosidosis (0.62 per 100,000). -
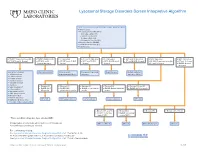
Lysosomal Storage Disorders Screen Interpretive Algorithm
Lysosomal Storage Disorders Screen Interpretive Algorithm LSDS / Lysosomal Storage Disorders Screen, Random, Urine Testing includes: ■ Mucopolysaccharides (MPS): – Dermatan sulfate (DS) – Heparan sulfate (HS) – Keratan sulfate (KS) – Chondroitin 6-sulfate (CS) ■ Oligosaccharides (OLIGO) ■ Ceramide trihexosides (CT) ■ Sulfatides (S) ■ OLIGO: Characteristic profile ■ OLIGO: MLII/III profile ■ S: Abnormal ■ CT and S: Abnormal ■ CT: Abnormal ■ MPS and S: Abnormal ■ MPS: Abnormal ■ MPS: Abnormal ■ CT, MPS and S: Normal ■ CT, MPS and S: ■ CT, MPS and OLIGO: ■ MPS and OLIGO: ■ MPS, OLIGO and S: ■ CT and OLIGO: Normal ■ OLIGO: Characteristic profile ■ CT and S: Normal Normal/abnormal Normal Normal Normal ■ CT and S: Normal ■ OLIGO: Normal/ abnormal One of the following: Mucolipidosis II/III Metachromatic Prosaposin/SaposinB Fabry Disease Multiple sulfatase ■ α-Mannosidosis leukodystrophy (MLD) deficiency deficiency (MSD) ■ β-Mannosidosis ■ Pompe disease ■ Sandhoff disease ■ Schindler disease ■ Sialidosis ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS ■ Elevated KS and CS ■ Galactosialidosis* ■ OLIGO: MPS ■ OLIGO: GM1 ■ OLIGO: α-Fucosidosis ■ OLIGO: Galactosialidosis ■ OLIGO: MPS IVA profile ■ α-Fucosidosis* IVB profile gangliosidosis profile profile profile ■ Mucolipidosis II/III* ■ GM1 gangliosidosis* ■ Morquio A & B* ■ NGYL1 deficiency MPS IVB GM1 gangliosidosis -Fucosidosis Galactosialidosis MPS IVA ■ MOGS-CDG (Congenital α Disorder of Glycosylation-IIb) ■ Elevated DS and HS ■ Elevated DS ■ Elevated HS ■ Elevated DS, HS, CS ■ OLIGO: -

Lysosomal Storage Disease Panel by Next-Generation Sequencing
TEST ID: LSDP LYSOSOMAL STORAGE DISEASE PANEL BY NEXT-GENERATION SEQUENCING USEFUL FOR REFERENCE VALUES } Follow up for abnormal biochemical results and confirmation of suspected lysosomal storage An interpretive report will be disease (LSD) provided. } Identifying mutations within genes known to be associated with lysosomal storage disease, allowing for predictive testing of at-risk family members ANALYTIC TIME 4 weeks CLINICAL INFORMATION Lysosomal storage diseases (LSDs) encompass a group of over 40 inherited biochemical diseases in which genetic mutations cause defective lysosomal functioning. Lysosomes perform catabolic functions for cells, which is accomplished through activity of various proteins such as lysosomal enzymes, transport proteins, and other proteins. Functional deficits in these proteins cause an accumulation of substrates in cells leading to progressive organ dysfunction. This leads to variable clinical features that can affect the cardiovascular, neurological, ocular, and skeletal systems, among others. Clinical features are dependent on the amount and location of the substrate accumulation, but may include the following: characteristic facial features (coarse features), hepatomegaly, deafness, vision loss, abnormal skeletal findings, hydrops fetalis, ataxia, hypotonia, developmental delay/regression, and intellectual disability. Age of onset is variable, with symptoms presenting from the prenatal period to adulthood, but generally LSDs are progressive and cause significant morbidity and mortality with a decreased lifespan. Enzyme replacement therapy and oral substrate inhibitors are therapeutic options for some LSDs. LSDs are inherited in an autosomal recessive manner with the exception of Hunter, Fabry, and Danon diseases, which are X-linked. There are some founder mutations associated with particular LSDs in the Ashkenazi Jewish and Finnish populations, leading to an increased carrier frequency for some.