Approach to Purpura Introduction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Urticarial Vasculitis Associated with Essential Thrombocythaemia
Acta Derm Venereol 2014; 94: 244–245 SHORT COMMUNICATION Urticarial Vasculitis Associated with Essential Thrombocythaemia Annabel D. Scott, Nicholas Francis, Helen Yarranton and Sarita Singh Dermatology Registrar, Department of Dermatology, Chelsea and Westminster Hospital, 369 Fulham Road, London SW10 9NH, United Kingdom. E-mail: [email protected] Accepted Apr 3, 2013; Epub ahead of print Aug 27, 2013 Urticarial vasculitis is a form of leucocytoclastic vas- prednisolone 30 mg once daily, which controlled her culitis whereby the skin lesions resemble urticaria. It is symptoms. associated with systemic lupus erythematosus, Sjögren’s A biopsy was taken from an uticarial lesion on the syndrome, hepatitis B and C viruses (1). Rarely it is asso- arm. Histology showed numerous perivascular neutro- ciated with an underlying haematological disorders and, phils and eosinophils with margination of neutrophils to the best of our knowledge, has never been reported in in the lumen of vessels and some leucocytoclasis with association with essential thrombocythaemia. We present red cell extravasation in keeping with an urticarial a case report of urticarial vasculitis associated with es- vasculitis (Fig. 2). sential thrombocythaemia. Blood tests revealed an erythrocyte sedimentation rate of 47 mm/h (normal range 1–12), a platelet count of 1,098 × 109/l (normal range 135–400) and an eosi- CASE REPORT nophilia of 1.0 × 109/l (normal range 0–0.2). Comple- A 32-year-old woman presented with a several month ment levels, thyroid function, serum iron, haemoglobin, history of recurrent urticaria without angioedema, 4 C-reactive protein, anti-nuclear antibody, anti-nuclear months post-partum. -

Appendix Search Strategy Treatment of Hemophilia.Pdf
Appendix Search strategies Hemophilia – general aspects PubMed (NLM) September 2009 Von Willebrand disease (TiAb) AND Controlled clinical trial (PT) NOT Purpura, Thrombocytopenic (Me) Angiohemophilia (TiAb) Meta analysis (PT) Blood coagulation disorders (Me) Randomized controlled trial (PT) Hemophilia (TiAb) Systematic (SB) Haemophilia (TiAb) Bleeding disorder (TiAb) Random* (Ti) Bleeding disorders (TiAb) OR Control* (Ti) NOT Medline (SB) ("controlled clinical trial"[Publication Type] OR "meta analysis"[Publication Type] OR "randomized controlled trial"[Publication Type] OR systematic[sb] OR ((random*[Title] OR control*[Title]) NOT Medline[sb])) AND ("von Willebrand Disease"[title/abstract] OR "angiohemophilia"[title/Abstract] OR "Blood Coagulation Disorders"[Mesh terms] OR "hemophilia"[title/abstract] OR "haemophilia"[title/abstract] OR "bleeding disorder"[title/abstract] OR "bleeding disorders"[Title/abstract]) NOT "Purpura, Thrombocytopenic"[MeSH Terms] 211 Hemophilia – general aspects Embase.com (Elsevier) September 2009 Blood clotting factor deficiency (Exp,MJR) AND Clinical trial (Exp) NOT Thrombocytopenic purpura (Exp) Von Willebrand disease (Ti) Intervention study (De) Angiohemophilia (Ti) Longitudinal study (De) Angiohaemophilia (Ti) Prospective study (De) Hemophilia (Ti) Meta analysis (De) Haemophilia (Ti) Systematic review (De) Bleeding disorder (Ti) Random* (Ti) Bleeding disorders (Ti) Control* (Ti) ('blood clotting factor deficiency'/exp/mjOR 'von willebrand disease':ti OR 'angiohemophilia':ti OR 'angiohaemophilia':ti OR 'hemophilia':ti -

Immune Thrombocytopenic Purpura with Subsequent Development of JAK2 V617F-Positive Essential Thrombocythemia: Case Report
ISSN: 2640-7914 DOI: https://dx.doi.org/10.17352/ahcrr CLINICAL GROUP Received: 13 July, 2021 Case Report Accepted: 03 August, 2021 Published: 04 August, 2021 *Corresponding author: Marisabel Hurtado-Castillo, Immune thrombocytopenic PGY-3, Internal Medicine, Department of Medicine at NYU, 536 Ovington Avenue Apartment 3, Brooklyn NYC 11209, USA, Tel: 718-312-9641; purpura with subsequent Email: Keywords: Immune thrombocytopenic purpura; development of JAK2 Essential thrombocythemia; JAK-STAT signaling path- way; JAK2(V617F) mutation V617F-positive essential https://www.peertechzpublications.com thrombocythemia: Case Report Marisabel Hurtado-Castillo1*, Brian Flaherty2 and Morris Jrada2 1PGY-3, Internal Medicine, Department of Medicine at NYU, Brooklyn, NY, USA 2Clinical Assistant Professor, Department of Medicine at NYU Grossman School of Medicine, Brooklyn, NY, USA Abstract The sequential occurrence of Immune Thrombocytopenic Purpura (ITP) and Essential Thrombocythemia (ET) has been reported in the literature on a few occasions, as these are two hematologic disorders with distinct etiologies and patients usually have contrasting clinical presentations. Our case highlights the sequential occurrence of ITP, followed by Janus kinase 2 (JAK2) (V617F)-positive ET in a 64-year-old white woman, after four years of follow-up. The pathophysiology relating to these two conditions is incompletely understood, however, JAK2(V617F) mutation has been found in all the cases reported. Early identifi cation of JAK2(V617F) mutation in a patient with a -

Thrombocytopenia.Pdf
THROMBOCYTOPENIA DIFFERENTIAL DIAGNOSIS FALSELY LOW PLATELET COUNT In vitro platelet clumping caused by EDTA-dependent agglutinins Giant platelets COMMON CAUSES OF THROMBOCYTOPENIA Pregnancy (gestational thrombocytopenia, preeclampsia) Drug-induced thrombocytopenia (i.e., heparin, quinidine, quinine, and sulfonamides) Viral infections (ie. HIV, rubella, infectious mononucleosis) Hypersplenism due to chronic liver disease Dilutional (massive transfusion) OTHER CAUSES OF THROMBOCYTOPENIA Myelodysplasia Congenital thrombocytopenia Thrombotic thrombocytopenic purpura (TTP) -hemolytic-uremic syndrome (HUS) Chronic disseminated intravascular coagulation (DIC) Autoimmune diseases, such as systemic lupus erythematosus-associated lymphoproliferative disorders (CLL and NHL) Sepsis Idiopathic thrombocytopenic purpura (ITP)* DIFFERENTIAL FOR THROMBOCYTOPENIA BASED ON CLINICAL SETTING CLINICAL SETTING DIFFERENTIAL DIAGNOSES Cardiac surgery Cardiopulmonary bypass, HIT, dilutional thrombocytopenia, PTP Interventional cardiac Abciximab or other IIb/IIIa blockers, HIT procedure Sepsis syndrome DIC, ehrlichiosis, sepsis, hemophagocytosis syndrome, drug-induced, misdiagnosed TTP, mechanical ventilation, pulmonary artery catheters Pulmonary failure DIC, hantavirus pulmonary syndrome, mechanical ventilation, pulmonary artery catheters Mental status TTP, ehrlichiosis changes/seizures Renal failure TTP, Dengue, HIT, DIC, HUS Continuous hemofiltration HIT, consumption by filter and tubing Cardiac failure HIT, drug-induced, pulmonary artery catheter Post-surgery -

Acute Immune Thrombocytopenic Purpura in Children
Turk J Hematol 2007; 24:41-51 REVIEW ARTICLE © Turkish Society of Hematology Acute immune thrombocytopenic purpura in children Abdul Rehman Sadiq Public School, Bahawalpur, Pakistan [email protected] Received: Sep 12, 2006 • Accepted: Mar 21, 2007 ABSTRACT Immune thrombocytopenic purpura (ITP) in children is usually a benign and self-limiting disorder. It may follow a viral infection or immunization and is caused by an inappropriate response of the immune system. The diagnosis relies on the exclusion of other causes of thrombocytopenia. This paper discusses the differential diagnoses and investigations, especially the importance of bone marrow aspiration. The course of the disease and incidence of intracranial hemorrhage are also discussed. There is substantial discrepancy between published guidelines and between clinicians who like to over-treat. The treatment of the disease ranges from observation to drugs like intrave- nous immunoglobulin, steroids and anti-D to splenectomy. The different modes of treatment are evaluated. The best treatment seems to be observation except in severe cases. Key Words: Thrombocytopenic purpura, bone marrow aspiration, Intravenous immunoglobulin therapy, steroids, anti-D immunoglobulins 41 Rehman A INTRODUCTION There is evidence that enhanced T-helper cell/ Immune thrombocytopenic purpura (ITP) in APC interactions in patients with ITP may play an children is usually a self-limiting disorder. The integral role in IgG antiplatelet autoantibody pro- American Society of Hematology (ASH) in 1996 duction -

Painless Purple Streaks on the Arms and Chest
PHOTO CHALLENGE Painless Purple Streaks on the Arms and Chest Tiffany Alexander, MD; Bernard Cohen, MD A 10-year-old boy presented with painless purple streaks on the arms and chest of 2 months’ duration. The rash recurred several times per month and cleared without treatment in 3 to 5 days. There was no history of trauma or medication exposure, and he was growing and developing normally.copy WHAT’S THE DIAGNOSIS? a. child maltreatment syndrome b. notfactitial purpura c. Henoch-Schönlein purpura d. idiopathic thrombocytopenic purpura Doe. meningococcemia PLEASE TURN TO PAGE E9 FOR THE DIAGNOSIS CUTIS Dr. Alexander was from the University of Maryland, Baltimore, and currently is from the Department of Dermatology, Duke University Medical Center, Durham, North Carolina. Dr. Cohen is from the Department of Dermatology, Division of Pediatric Dermatology, Johns Hopkins University School of Medicine, Baltimore. The authors report no conflict of interest. Correspondence: Bernard Cohen, MD, Johns Hopkins University School of Medicine, Division of Pediatric Dermatology, David M. Rubenstein Child Health Bldg, Ste 2107, 200 N Wolfe St, Baltimore, MD 21287 ([email protected]). E8 I CUTIS® WWW.MDEDGE.COM/DERMATOLOGY Copyright Cutis 2019. No part of this publication may be reproduced, stored, or transmitted without the prior written permission of the Publisher. PHOTO CHALLENGE DISCUSSION THE DIAGNOSIS: Factitial Purpura actitial dermatologic disorders are characterized by the state of one’s health and life span.4 Cupping is per- skin findings triggered by deliberate manipulation formed by placing a glass cup over a painful body part. F of the skin with objects to create lesions and feign A partial vacuum is created by flaming, mechanical with- signs of a dermatologic condition to seek emotional drawal, or thermal cooling of the entrapped air under the and psychological benefit.1 The etiology of the lesions is cup. -

Immune Thrombocytopenic Purpura (ITP) — Adult Conditions for Which Ivig Has an Established Therapeutic Role
Immune thrombocytopenic purpura (ITP) — adult Conditions for which IVIg has an established therapeutic role. Specific Conditions Newly Diagnosed Immune thrombocytopenic purpura (ITP) Persistent Immune thrombocytopenic purpura (ITP) Chronic Immune thrombocytopenic purpura (ITP) Evans syndrome ‐ with significant Immune thrombocytopenic purpura (ITP) ‐ adult Indication for IVIg Use Newly diagnosed ITP — initial Ig therapy ITP in pregnancy — initial Ig therapy ITP with life‐threatening haemorrhage or the potential for life‐threatening haemorrhage Newly diagnosed or persistent ITP — subsequent therapy (diagnosis <12 months) Refractory persistent or chronic ITP — splenectomy failed or contraindicated and second‐line agent unsuccessful Subsequent or ongoing treatment for ITP responders during pregnancy and the postpartum period ITP and inadequate platelet count for planned surgery HIV‐associated ITP Level of Evidence Evidence of probable benefit – more research needed (Category 2a) Description and Diagnostic Immune thrombocytopenic purpura (ITP) is a reduction in platelet count Criteria (thrombocytopenia) resulting from shortened platelet survival due to anti‐platelet antibodies, reduced platelet production due to immune induced reduced megakaryopoeisis and/or immune mediated direct platelet lysis. When counts are very low (less than 30x109/L), bleeding into the skin (purpura) and mucous membranes can occur. Bone marrow platelet production (megakaryopoiesis) is morphologically normal. In some cases, there is additional impairment of platelet function related to antibody binding to glycoproteins on the platelet surface. It is a common finding in patients with human immunodeficiency virus (HIV) disease, and while it may be found at any stage of the infection, its prevalence increases as HIV disease advances. Around 80 percent of adults with ITP have the chronic form of disease. -

Thrombocytopenia in Older Adults Melissa (Kah Poh) Loh, MD
URMC Division of Geriatrics & Aging March 2019 Thrombocytopenia in Older adults Melissa (Kah Poh) Loh, MD Background Thrombocytopenia (or low platelet count) is a common hematologic abnormality encountered in older adults Common causes of thrombocytopenia include (Table 1): Immune-related [e.g. immune thrombocytopenia purpura (ITP)] Drug-induced [e.g. heparin-induced thrombocytopenia (HIT)] Bone marrow failure Other: Infections (e.g. H. pylori, HIV, hepatitis), pseudothrombocytopenia (Figure 1), alcohol abuse, liver Figure 1: Peripheral smear showing clumped cirrhosis, blood transfusion, thrombotic thrombocytopenia platelets (pseudothrombocytopenia — falsely purpura (TTP), and hemolytic-uremic syndrome (HUS), low platelet count) thyroid disease Age-related changes in the organ and vasculature systems increase the risks of thrombocytopenia on hemostasis Factors that may enhance bleeding in older adults. These include: Comorbidities Medications Loss of subcutaneous tissue Age-specific factors that may prevent bleeding in older adults include enhanced platelet aggregation and increased fibrinogen, factor V, and von Willebrand factor Thrombocytopenia is often multifactorial in older adults. Table 1: Quick Facts on Thrombocytopenia PATHOPHYSIOLOGY Immune Thrombocytopenia Purpura Drug-Induced Increased platelet clearance in the reticulo-endothelial Impaired production (direct marrow toxicity or system of bone marrow, spleen, and/or liver megakaryocyte-specific inhibition) Inadequate platelet production due to megakaryocyte Increased platelet clearance (indirect immune clearance, inhibition by IgG antibodies antibody-specific immune clearance, miscellaneous immune -mediated) Often suppress other cell lines CAUSES Immune Thrombocytopenia Purpura Drug-Induced Often idiopathic See Table 2 Can be associated with underlying hematologic abnormalities (e.g. myelodysplastic syndrome, chronic lymphocytic leukemia) INCIDENCE RATES Immune Thrombocytopenia Purpura Drug-Induced 4.62 per 100,000 adults aged >60 years vs. -

Essential Thrombocythaemia
Andrews_EU Onc & Haema 09/05/2011 15:19 Page 125 Essential Thrombocythaemia Platelets – From Function to Dysfunction in Essential Thrombocythaemia Robert K Andrews,1 Michael C Berndt2 and Ismael Elalamy3 1. Associate Professor, Australian Centre for Blood Diseases, Monash University; 2. Director, Biomedical Diagnostic Institute, Dublin City University; 3. Professor, Haematology Department, Hôpital Tenon Paris University VI Abstract Platelets are an important component of blood. The main biological role of platelets is to respond to vascular injury and promote thrombus formation to prevent bleeding. However, we now know that platelets also have additional functions in a variety of processes such as immunity, inflammation, coagulation, atherogenesis and tumour metastasis. Platelet disorders commonly lead to defects in haemostasis. Of particular interest is the myeloid proliferative disorder, essential thrombocythaemia (ET). In ET the increased number of platelets leads to an increased risk of blood clot formation and subsequent thrombohaemorrhagic complications. Here we provide a general review of platelet function and activation, as well as more detailed information on the dysfunction of platelets in patients with ET. Keywords Haemostasis, megakaryocytes, platelets, platelet adhesion, platelet aggregation, platelet receptors Disclosure: The authors have no conflicts of interest to declare. Acknowledgements: The authors directed writing assistance from Ewen Buckling, an employee of iMed Comms. Editorial assistance in the form of proofreading, copy editing and fact checking was also provided by iMed Comms. iMed Comms was funded by Shire Pharmaceuticals for support in writing and editing this manuscript. This article was conceived at a Shire-sponsored scientific advisory board, at which Michael C Berndt and Ismael Elalamy were attendees and were recompensed for their attendance. -

Henoch-Schönlein-Purpura-Fact-Sheet.Pdf
PATIENT FACT SHEET Henoch Schönlein Purpura (HSP) Henoch Schönlein Purpura (HSP) is a type of vasculitis, bleeding into the skin, which causes the rash of HSP. which means inflammation of the blood vessels. The Bleeding into the stool and urine can also occur. In North exact cause of HSP is unknown and there is no specific America, HSP is the most common form of vasculitis in test to diagnose. children. It is most common in ages 3 to 15 years and is more common in boys than girls. It rarely occurs in adults. In patients with HSP, the person’s own immune system CONDITION HSP can occur at any time of the year, though it is more attacks blood vessels in the skin, intestines, joints and common in the winter. DESCRIPTION kidneys. Inflammation in the blood vessel wall leads to The classic symptom of HSP is a red to dark purple rash, up to one-third of patients can develop kidney disease. called “purpura”, which is often most severe on the legs Treatment depends on the individual child’s symptoms. and buttocks. Other symptoms include painful swelling Monitoring for kidney disease plays an important role in around the joints, swelling under the skin and abdominal early detection and treatment if this complication happens. pain. Many children with HSP recover completely, but SIGNS/ SYMPTOMS Most children with HSP do not require any specific One of the most important parts of HSP management is treatment and recover with time alone. Joint pain can often monitoring for kidney disease. Temporary blood or protein be controlled with rest and over-the-counter medications, in the urine is common. -

Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management
Journal of Clinical Medicine Review Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management Senthil Sukumar 1 , Bernhard Lämmle 2,3,4 and Spero R. Cataland 1,* 1 Division of Hematology, Department of Medicine, The Ohio State University, Columbus, OH 43210, USA; [email protected] 2 Department of Hematology and Central Hematology Laboratory, Inselspital, Bern University Hospital, University of Bern, CH 3010 Bern, Switzerland; [email protected] 3 Center for Thrombosis and Hemostasis, University Medical Center, Johannes Gutenberg University, 55131 Mainz, Germany 4 Haemostasis Research Unit, University College London, London WC1E 6BT, UK * Correspondence: [email protected] Abstract: Thrombotic thrombocytopenic purpura (TTP) is a rare thrombotic microangiopathy charac- terized by microangiopathic hemolytic anemia, severe thrombocytopenia, and ischemic end organ injury due to microvascular platelet-rich thrombi. TTP results from a severe deficiency of the specific von Willebrand factor (VWF)-cleaving protease, ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, member 13). ADAMTS13 deficiency is most commonly acquired due to anti-ADAMTS13 autoantibodies. It can also be inherited in the congenital form as a result of biallelic mutations in the ADAMTS13 gene. In adults, the condition is most often immune-mediated (iTTP) whereas congenital TTP (cTTP) is often detected in childhood or during pregnancy. iTTP occurs more often in women and is potentially lethal without prompt recognition and treatment. Front-line therapy includes daily plasma exchange with fresh frozen plasma replacement and im- munosuppression with corticosteroids. Immunosuppression targeting ADAMTS13 autoantibodies Citation: Sukumar, S.; Lämmle, B.; with the humanized anti-CD20 monoclonal antibody rituximab is frequently added to the initial ther- Cataland, S.R. -
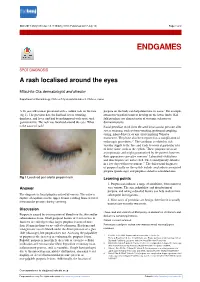
A Rash Localised Around the Eyes
BMJ 2017;358:j3148 doi: 10.1136/bmj.j3148 (Published 2017 July 13) Page 1 of 2 Endgames ENDGAMES SPOT DIAGNOSIS A rash localised around the eyes Mitsuhito Ota dermatologist and director Department of Dermatology, Chitose City Hospital Hokkou 2, Chitose, Japan A 26 year old woman presented with a sudden rash on the face purpura on the body can help determine its cause.1 For example, (fig 1). The previous day, she had had severe vomiting, cutaneous vasculitis tends to develop on the lower limbs. Nail diarrhoea, and fever, and had been diagnosed with acute viral fold petechiae are characteristic of systemic sclerosis or gastroenteritis. The rash was localised around the eyes. What dermatomyositis. is the cause of rash? Facial petechiae result from elevated intravascular pressure after severe straining, such as from vomiting, prolonged coughing, crying, infant delivery, or any effort implying Valsalva manoeuvre. They have also been reported as a complication of endoscopic procedures.2 3 The condition is related to rich vascular supply to the face and tends to occur at particular sites of loose tissue, such as the eyelids.4 These purpuric areas are asymptomatic and might go unnoticed by the patient; however, their appearance can cause concern.5 Laboratory evaluations and skin biopsies are not needed. The lesion typically subsides in a few days without treatment.2-4 The differential diagnoses of purpura locally on the eyelids include amyloidosis associated purpura (panda sign) and purpura related to neuroblastoma. Fig 1 Localised peri-orbital pinpoint rash Learning points 1. Purpura can indicate a range of conditions, from minor to Answer very serious.