ABSTRACT ALLEN, JOSHUA LEE. Molecular Interactions of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(12) United States Patent (10) Patent No.: US 7.449,161 B2 Boryta Et Al
USOO74491.61B2 (12) United States Patent (10) Patent No.: US 7.449,161 B2 Boryta et al. (45) Date of Patent: Nov. 11, 2008 (54) PRODUCTION OF LITHIUM COMPOUNDS 4,207,297 A * 6/1980 Brown et al. ............. 423,179.5 DIRECTLY FROM LITHIUM CONTAINING 4,243,641 A * 1/1981 Ishimori et al. .......... 423/179.5 BRINES 4.261,960 A * 4/1981 Boryta ............. ... 423,179.5 4,271,131. A * 6/1981 Brown et al. ...... ... 423,179.5 4,274,834. A 6/1981 Brown et al. .............. 23,302 R (75) Inventors: Daniel Alfred Boryta, Cherryville, NC 4,463,209 A * 7/1984 Kursewicz et al. .......... 585/467 (US); Teresita Frianeza Kullberg, 4,465,659 A * 8/1984 Cambridge et al. ......... 423,495 Gastonia, NC (US); Anthony Michael 4,588,565 A * 5/1986 Schultze et al. .......... 423,179.5 Thurston, Edmond, OK (US) 4,747.917 A * 5/1988 Reynolds et al. ............ 205,512 4,859,343 A * 8/1989 Frianeza-Kullberg (73) Assignee: Chemetall Foote Corporation, Kings et al. .......................... 210,679 Mountain, NC (US) 4,980,136 A * 12/1990 Brown et al. ... 423,179.5 5,049,233 A 9, 1991 Davis .......................... 216.93 (*) Notice: Subject to any disclaimer, the term of this 5,219,550 A * 6/1993 Brown et al. ............. 423 (419.1 patent is extended or adjusted under 35 5,599,516 A * 2/1997 Bauman et al. .......... 423/179.5 U.S.C. 154(b) by 0 days. 5,939,038 A * 8/1999 Wilkomirsky ............... 423,276 5.993,759 A * 1 1/1999 Wilkomirsky ........... -

The Secondary Phosphate Minerals from Conselheiro Pena Pegmatite District (Minas Gerais, Brazil): Substitutions of Triphylite and Montebrasite Scholz, R.; Chaves, M
The secondary phosphate minerals from Conselheiro Pena Pegmatite District (Minas Gerais, Brazil): substitutions of triphylite and montebrasite Scholz, R.; Chaves, M. L. S. C.; Belotti, F. M.; Filho, M. Cândido; Filho, L. Autor(es): A. D. Menezes; Silveira, C. Publicado por: Imprensa da Universidade de Coimbra URL persistente: URI:http://hdl.handle.net/10316.2/31441 DOI: DOI:http://dx.doi.org/10.14195/978-989-26-0534-0_27 Accessed : 2-Oct-2021 20:21:49 A navegação consulta e descarregamento dos títulos inseridos nas Bibliotecas Digitais UC Digitalis, UC Pombalina e UC Impactum, pressupõem a aceitação plena e sem reservas dos Termos e Condições de Uso destas Bibliotecas Digitais, disponíveis em https://digitalis.uc.pt/pt-pt/termos. Conforme exposto nos referidos Termos e Condições de Uso, o descarregamento de títulos de acesso restrito requer uma licença válida de autorização devendo o utilizador aceder ao(s) documento(s) a partir de um endereço de IP da instituição detentora da supramencionada licença. Ao utilizador é apenas permitido o descarregamento para uso pessoal, pelo que o emprego do(s) título(s) descarregado(s) para outro fim, designadamente comercial, carece de autorização do respetivo autor ou editor da obra. Na medida em que todas as obras da UC Digitalis se encontram protegidas pelo Código do Direito de Autor e Direitos Conexos e demais legislação aplicável, toda a cópia, parcial ou total, deste documento, nos casos em que é legalmente admitida, deverá conter ou fazer-se acompanhar por este aviso. pombalina.uc.pt digitalis.uc.pt 9 789892 605111 Série Documentos A presente obra reúne um conjunto de contribuições apresentadas no I Congresso Imprensa da Universidade de Coimbra Internacional de Geociências na CPLP, que decorreu de 14 a 16 de maio de 2012 no Coimbra University Press Auditório da Reitoria da Universidade de Coimbra. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0233042 A1 Boryta Et Al
US 20080233042A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0233042 A1 Boryta et al. (43) Pub. Date: Sep. 25, 2008 (54) PRODUCTION OF LITHIUM COMPOUNDS 11/332,134, filed on Jan. 13, 2006, now Pat. No. 7,214, DIRECTLY FROM LITHIUM CONTAINING 355, which is a division of application No. 10/395,984, BRINES filed on Mar. 25, 2003, now Pat. No. 7,157,065, which is a continuation-in-part of application No. 09/707, (76) Inventors: Daniel Alfred Boryta, Cherryville, 427, filed on Nov. 7, 2000, now Pat. No. 6,936,229, NC (US); Teresita Frianeza which is a division of application No. 09/353,185, filed Kullberg, Gastonia, NC (US); on Jul. 14, 1999, now Pat. No. 6,207,126. Anthony Michael Thurston, Edmond, OK (US) Publication Classification Correspondence Address: (51) Int. C. FULBRIGHT & JAWORSKI, LLP C22B 26/10 (2006.01) 666 FIFTHAVE NEW YORK, NY 10103-3198 (US) (52) U.S. Cl. ..................................................... 423/499.3 (21) Appl. No.: 12/114,054 (57) ABSTRACT (22) Filed: May 2, 2008 Methods and apparatus for the production of low sodium lithium carbonate and lithium chloride from a brine concen Related U.S. Application Data trated to about 6.0 wt % lithium are disclosed. Methods and (60) Continuation of application No. 1 1/706027, filed on apparatus for direct recovery of technical grade lithium chlo Feb. 14, 2007, which is a division of application No. ride from the concentrated brine are also disclosed. Patent Application Publication Sep. 25, 2008 Sheet 1 of 7 US 2008/0233042 A1 LITHIUM CARBONATE PROCESSFLOW DIAGRAM (A) 2 CD SOLID SODAASH 4 3 Mg MUD \ SAS 6 FROM C5 5 REACTOR LIQUOR MOTHER LIOUOR WASH FILTER (10) BRINE + WASHFILTRATE 0.5 - 1.2% Li 2ND STAGE 12 REACTOR FILTER Li2CO3 REACTOR 18 FILTER MOTHER LIQUOR (19) WASH WATER FLTRATE PROCESS BLEED WET LITHIUM CARBONATE9 21 DRYER WET Li2CO3 22 TO B1 TECH GRADE Li2CO3 FIG. -

Local Peralkalinity in Peraluminous Granitic Pegmatites. I. Evidence
This is the peer-reviewed, final accepted version for American Mineralogist, published by the Mineralogical Society of America. The published version is subject to change. Cite as Authors (Year) Title. American Mineralogist, in press. DOI: https://doi.org/10.2138/am-2021-7790. http://www.minsocam.org/ Revision 1 1 Local peralkalinity in peraluminous granitic pegmatites. I. Evidence 2 from whewellite and hydrogen carbonate in fluid inclusions 3 YONGCHAO LIU1,2, CHRISTIAN SCHMIDT2,*, AND JIANKANG LI1 4 1MNR Key Laboratory of Metallogeny and Mineral Assessment, Institute of Mineral Resources, 5 Chinese Academy of Geological Sciences, Beijing 100037, China 6 2GFZ German Research Centre for Geosciences, Telegrafenberg, 14473 Potsdam, Germany 7 *Corresponding author: [email protected] 8 9 ABSTRACT 10 Fluid inclusions in pegmatite minerals were studied using Raman spectroscopy to 11 determine the carbon species. Carbon dioxide is very abundant in the aqueous liquid and vapor − 12 phases. Occasionally, CH4 was found in the vapor. In the aqueous liquid, HCO3 was detected in 13 fluid inclusions in tantalite-(Mn) from the Morrua Mine and in late-stage quartz from the Muiâne 14 pegmatite and the Naipa Mine, all in the Alto Ligonha District, Mozambique. Moreover, we 15 observed a carbonate (calcite group) in fluid inclusions in garnet from the Naipa Mine and in 16 beryl from the Morrua Mine, both in the Alto Ligonha District, Mozambique, and a calcite-group 17 carbonate and whewellite [CaC2O4∙H2O] in fluid inclusions in topaz from Khoroshiv, Ukraine. 18 The occurrence of oxalate is interpreted to be due to a reaction of some form of carbon (possibly 19 CO or bitumen) with a peralkaline fluid. -

COMPUTER SIMULATION STUDIES of ... -.:University of Limpopo
COMPUTER SIMULATION STUDIES OF SPINEL LiMn2O4 AND SPINEL LiNiXMn2-XO4 (0≤x≤2) By KEMERIDGE TUMELO MALALTJI Thesis Submitted in fulfilment of the requirements for the degree of Doctor of Philosophy In Physics in the FACULTY OF SCIENCES AND AGRICULTURE (School of Physical and Mineral Sciences) at the UNIVERSITY OF LIMPOPO SUPERVISOR: Prof. R.R. Maphanga CO- SUPERVISORS: Prof. P.E. Ngoepe (Dated: 29 October 2019) Declaration I, Kemeridge Tumelo Malatji confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. I agree that the Library may lend or copy this dissertation on request. Signature: Date: 29/10/2019 Name: Mr K.T. Malatji ii Abstract LiMn2O4 spinel (LMO) is a promising cathode material for secondary lithium-ion batteries which, despite its high average voltage of lithium intercalation, suffers crystal symmetry lowering due to the Jahn-Teller active six-fold Mn3+ cations. Although Ni has been proposed as a suitable substitutional dopant to improve the energy density of LiMn2O4 and enhance the average lithium intercalation voltage, the thermodynamics of Ni incorporation and its effect on the electrochemical properties of this spinel are not fully understood. Firstly, structural, electronic and mechanical properties of spinel LiMn2O4 and LiNixMn2-xO4 have been calculated out using density functional theory employing the pseudo-potential plane-wave approach within the generalised gradient approximation, together with Virtual Cluster Approximation. The structural properties included equilibrium lattice parameters; electronic properties cover both total and partial density of states and mechanical properties investigated elastic properties of all systems. -

The Journal of ^ Y Volume 26 No
Gemmolog^^ The Journal of ^ y Volume 26 No. 8 October 1999 fj J The Gemmological Association and Gem Testing Laboratory of Great Britain Gemmological Association and Gem Testing Laboratory of Great Britain 27 Greville Street, London EC1N 8TN Tel: 020 7404 3334 Fax: 020 7404 8843 e-mail: [email protected] Website: www.gagtl.ac.uk/gagtl. President: Professor R.A. Howie Vice-Presidents: E.M. Bruton, A.E. Farn, D.G. Kent, R.K. Mitchell Honorary Fellows: Chen Zhonghui, R.A. Howie, R.T. Liddicoat Jnr, K. Nassau Honorary Life Members: H. Bank, D.J. Callaghan, E.A. Jobbins, H. Tillander Council of Management: T.J. Davidson, N.W. Deeks, R.R. Harding, I. Mercer, J. Monnickendam, M J. O'Donoghue, E. Stern, I. Thomson, V.P. Watson Members' Council: A.J. Allnutt, P. Dwyer-Hickey, S.A. Everitt, A.G. Good, J. Greatwood, B. Jackson, L. Music, J.B. Nelson, PG. Read, R. Shepherd, P.J. Wates, C.H. Winter Branch Chairmen: Midlands - G.M. Green, North West -1. Knight, Scottish - B. Jackson Examiners: A.J. Allnutt, MSc, Ph.D., FGA, L. Bartlett, B.Sc, MPhiL, FGA, DGA, E.M. Bruton, FGA, DGA, S. Coelho, B.Sc, FGA, DGA, Prof. A.T. Collins, B.Sc, Ph.D, A.G. Good, FGA, DGA, J. Greatwood, FGA, G.M. Howe, FGA, DGA, B. Jackson, FGA, DGA, G.H. Jones, B.Sc, Ph.D., FGA, M. Newton, B.Sc, D.Phil., C.J.E. Oldershaw, B.Sc (Hons), FGA, H.L. Plumb, B.Sc, FGA, DGA, R.D. Ross, B.Sc, FGA, DGA, PA. -

Standard X-Ray Diffraction Powder Patterns NATIONAL BUREAU of STANDARDS
NBS MONOGRAPH 25—SECTION 1 9 CO Q U.S. DEPARTMENT OF COMMERCE/National Bureau of Standards Standard X-ray Diffraction Powder Patterns NATIONAL BUREAU OF STANDARDS The National Bureau of Standards' was established by an act of Congress on March 3, 1901. The Bureau's overall goal is to strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to promote public safety. The Bureau's technical work is per- formed by the National Measurement Laboratory, the National Engineering Laboratory, and the Institute for Computer Sciences and Technology. THE NATIONAL MEASUREMENT LABORATORY provides the national system of physical and chemical and materials measurement; coordinates the system with measurement systems of other nations and furnishes essentia! services leading to accurate and uniform physical and chemical measurement throughout the Nation's scientific community, industry, and commerce; conducts materials research leading to improved methods of measurement, standards, and data on the properties of materials needed by industry, commerce, educational institutions, and Government; provides advisory and research services to other Government agencies; develops, produces, and distributes Standard Reference Materials; and provides calibration -

(12) United States Patent (10) Patent No.: US 8,722,004 B2 Wu Et Al
USOO8722004B2 (12) United States Patent (10) Patent No.: US 8,722,004 B2 Wu et al. (45) Date of Patent: May 13, 2014 (54) METHOD FOR THE PREPARATION OFA 6,730,281 B2 * 5/2004 Barker et al. ................. 423.306 LITHIUM PHOSPHATE COMPOUND WITH 2006/0147365 A1* 7, 2006 Okada et al. ... ... 423.306 2006/0257307 A1* 11/2006 Yang ........... ... 423.306 AN OLIVINE CRYSTAL STRUCTURE 2008, 0008938 A1* 1/2008 Wu et al. ... 429,221 2010.0059.706 A1 3/2010 Dai et al. ................... 252, 1821 (75) Inventors: Mark Y. Wu, Wujie Township, Yilan County (TW); Cheng-Yu Hsieh, Wujie FOREIGN PATENT DOCUMENTS Township, Yilan County (TW); Chih-Hao Chiu, Wujie Township, Yilan TW I254.031 5, 2006 County (TW) TW I29263.5 1, 2008 (73) Assignee: Phosage, Inc., Yilan County (TW) OTHER PUBLICATIONS (*) Notice: Subject to any disclaimer, the term of this Shiraishi et al., “Formation of impurities on phospho-olivine patent is extended or adjusted under 35 LiFePO4 during hydrothermal synthesis,” Journal of Power Sources, U.S.C. 154(b) by 710 days. 2005, pp. 555-558, vol. 146. (21) Appl. No.: 12/478,387 * cited by examiner (22) Filed: Jun. 4, 2009 Primary Examiner — Melvin C Mayes (65) Prior Publication Data Assistant Examiner — Colette Nguyen US 2010/O2O2951A1 Aug. 12, 2010 (74) Attorney, Agent, or Firm — Muncy, Geissler, Olds & Lowe, P.C. (30) Foreign Application Priority Data (57) ABSTRACT Feb. 12, 2009 (TW) ............................... 98104490 A The present invention relates to a method for the preparation (51) Int. Cl. of a lithium phosphate compound with an olivine crystal COB 25/26 (2006.01) structure, which has a chemical formula of Li, M.M'PO, (52) U.S. -

Technological Gaps Inhibiting the Exploitation of Crms Primary Resources
SCRREEN Coordination and Support Action (CSA) This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 730227. Start date : 2016-11-01 Duration : 38 Months www.scrreen.eu Technological gaps inhibiting the exploitation of CRMs primary resources Authors : Dr. Jordi GUIMERA (AMPHOS21) SCRREEN - D6.1 - Issued on 2019-12-18 22:10:32 by AMPHOS21 SCRREEN - D6.1 - Issued on 2019-12-18 22:10:32 by AMPHOS21 SCRREEN - Contract Number: 730227 Project officer: Jonas Hedberg Document title Technological gaps inhibiting the exploitation of CRMs primary resources Author(s) Dr. Jordi GUIMERA Number of pages 126 Document type Deliverable Work Package WP6 Document number D6.1 Issued by AMPHOS21 Date of completion 2019-12-18 22:10:32 Dissemination level Public Summary Technological gaps inhibiting the exploitation of CRMs primary resources Approval Date By 2019-12-19 10:05:27 Mrs. Maria TAXIARCHOU (LabMet, NTUA) 2020-01-27 08:01:27 Mr. Stéphane BOURG (CEA) SCRREEN - D6.1 - Issued on 2019-12-18 22:10:32 by AMPHOS21 SCRREEN This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 730227. Start date: 2016-11-01 Duration: 37 Months DELIVERABLE 6.1: TECHNOLOGICAL GAPS AND BARRIERS ON PRIMARY RESOURCES AUTHOR(S): Aina Bruno Diaz (Amphos 21) Susanna Casanovas (Amphos 21) Lena Sundqvist (SWERIM) Stéphane Bourg (CEA) Michael Samouhos (NTUA) DATE OF SUBMISSION: 09/12/2019 CONTENT List of figures .......................................................................................................................................................... -
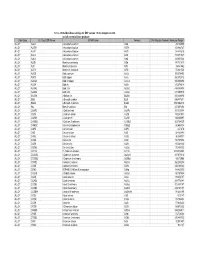
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

The Microscopic Determination of the Nonopaque Minerals
DEPARTMENT OF THE INTERIOR ALBERT B. FALL, Secretary UNITED STATES GEOLOGICAL SURVEY GEORGE OTIS SMITH, Director Bulletin 679 THE MICROSCOPIC DETERMINATION OF THE NONOPAQUE MINERALS BY ESPER S. LARSEN WASHINGTON GOVERNMENT PRINTING OFFICE 1921 CONTENTS. CHAPTER I. Introduction.................................................. 5 The immersion method of identifying minerals........................... 5 New data............................................................. 5 Need of further data.................................................... 6 Advantages of the immersion method.................................... 6 Other suggested uses for the method.................................... 7 Work and acknowledgments............................................. 7 CHAPTER II. Methods of determining the optical constants of minerals ....... 9 The chief optical constants and their interrelations....................... 9 Measurement of indices of refraction.................................... 12 The embedding method............................................ 12 The method of oblique illumination............................. 13 The method of central illumination.............................. 14 Immersion media.................................................. 14 General features............................................... 14 Piperine and iodides............................................ 16 Sulphur-selenium melts....................................... 38 Selenium and arsenic selenide melts........................... 20 Methods of standardizing -

The Composition and Distribution of Phosphate Rock with Special Reference to the United States
~g 2 8 11111 . 1/11/2.5 2 5 ~g. ~~ IIIF8 11111 . 1.0 3 2 1.0 B;;;, 11111 . !;;j 1I111~J, 2;..; 2.2 !.;. 1'111361.1- :: IIIII~ 1'- I:" .... }9~ '- :: ;4,& 2.0 1.1 ...........'- - 1.1 !:...:: 11 111111.8 111111.25 111111.4 '""1.6 111111.25 11111_1.4 "'" 1.6 MICROCOPY RESOLUT'DN WiT CHART MICROCOPY RlSOLUTION TEST CHART NATIONAL BUREAU (1F SlANfJARDo 1%, A ~==~~==-~~~~====~==~~ TECHNICAL Bl'1.LETINNO.364~ JUNE 1933 UNITED STATES DEPARTMENT OF AGRICULTURE WASHING""'ON, D. C THE COMPOSITION AND DISTRIBUTION OF PHOS PHATE ROC ~~ WITH SPECIAL REFEREHCE TO THE UNITED STATES By K. D. JACOB, Chemist, and W. L. TrILL, H. L. MARSHALL, and D. S. REY NOJ.DS, Assill/ant Chem':sts, Division oj Fertiliz~ Technology, Fertili,~er and Fixed Nitrogen Investigations, Bu,real.<. of Chemistry and Soils.' CONTENT!'. Page Page Introductlou________________________________ 1 Methods of chemlc.nl analysis________________ ]7 ReviewStates _______________________of phosphate deposits •_of .the __________ Uuited _ Results of chemical analyses_._______________ 19 4 Pbosphoric acid __ •______ ._______________ 25 Alabama____________ .___ ._______________ _ 26 Arkansas_______________________________ _ 4 o.ilcium and magnesium________________ 4 .5Juminum, iron, and titanium••________ 26 Florlda_____ •_________ •_. ____ . __________ _ 4 Sodium and potassium________..________ 27 6 :Manganese, chromium, and vanadium__ 27 Copper, zinc, and arsenlc__________ ._ ____ 29 Illinois~e~~~~~:=::=::::::__________ •_____:::= •::::::::~:::::::::____ • __ ..________ 6 Kentucky__ ,___ ._.____ •___ •• __ •_________ 6 Barium,elements rare_______________________ earths, and other metallic•_______ 6 Silica and carbon dloxide _____• __________ 31 r. Fluorine_________ •________ •__________ •___ 32 32 Chlorine, brQmlne, and iodine._________ _ g:~~!~~~e:~~=~::=::::::::::~::::::::~::Nevada_________ ._._.__ •_______ ....