Chem365 Labbook-2021
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Exam 1 (February 23, 2004) ID# ______
Chemistry 211 Name ___________________________ Exam 1 (February 23, 2004) ID# ___________________________ 10 1. You desire to synthesize 3-ethyl-3-pentanol starting with an ester. (i) What would be the name of the ester, and what is the name for the Grignard reagent (e.g., methyl magnesium bromide)? (ii) For the carbons shown in the product, show plausible hydrocarbons that you could start with to produce the ester and the Grignard reagent (as in a retrosynthesis). 12 2. (i) Show the step-by-step process required to produce propyllithium, which requires a free radical reaction mechanism, . (ii) Show the complete reaction mechanism for reaction between propyllithium and the correct ketone to produce 3-propyl-3-pentanol. (iii) Propose a possible reaction mechanism by which dipropyl cuprate (Cu+ with two propyl groups attached) could react with ethyl bromide to produce a new hydrocarbon. (This is a thinking exercise! So, think! () 8 3. As mentioned in the text, diethyl ether, pentane, and 1-butanol have similar molar masses, but different physical properties. Boiling points are 35oC, 36oC, and 117oC, respectively. Their respective solubilities in water are 7.5g/100mL, insoluble, and 9g/100mL. (i) Draw structures for each of these compounds. (ii) Justify the observed boiling points and their solubilities. 16 4. Draw structures of the following compounds 2,3-heptanediol isopropyllithium benzylmagnesium bromide benzoic acid benzaldehylde dimethyl sulfide t-butyl methanoate dibutyl ketone 12 5. Alcohols can be oxidized to produce other compounds, and can be produced by reduction. For the reactions shown below, show the structure for the expected product (if reaction does not occur, state: No Reaction) when treated with the indicated oxidizing or reducing agents. -

Grignard Reaction: Synthesis of Triphenylmethanol
*NOTE: Grignard reactions are very moisture sensitive, so all the glassware in the reaction (excluding the work-up) should be dried in an oven with a temperature of > 100oC overnight. The following items require oven drying. They should be placed in a 150mL beaker, all labeled with a permanent marker. 1. 5mL conical vial (AKA: Distillation receiver). 2. Magnetic spin vane. 3. Claisen head. 4. Three Pasteur pipettes. 5. Two 1-dram vials (Caps EXCLUDED). 6. One 2-dram vial (Caps EXCLUDED). 7. Glass stirring rod 8. Adaptor (19/22.14/20) Grignard Reaction: Synthesis of Triphenylmethanol Pre-Lab: In the “equations” section, besides the main equations, also: 1) draw the equation for the production of the byproduct, Biphenyl. 2) what other byproduct might occur in the reaction? Why? In the “observation” section, draw data tables in the corresponding places, each with 2 columns -- one for “prediction” (by answering the following questions) and one for actual drops or observation. 1) How many drops of bromobenzene should you add? 2) How many drops of ether will you add to flask 2? 3) 100 µL is approximately how many drops? 4) What are the four signs of a chemical reaction? (Think back to Chem. 110) 5) How do the signs of a chemical reaction apply to this lab? The Grignard reaction is a useful synthetic procedure for forming new carbon- carbon bonds. This organometallic chemical reaction involves alkyl- or aryl-magnesium halides, known as Grignard 1 reagents. Grignard reagents are formed via the action of an alkyl or aryl halide on magnesium metal. -

Organic Chemistry Laboratory II Preparation of Triphenylmethanol (Grignard Reaction) Experiment Procedure (Printable Pdf Format)
grigdes http://www.organicchem.org/oc2web/lab/exp/grig/grigdesbenzophenone... Organic Chemistry II | Lecture | Laboratory Organic Chemistry Laboratory II Preparation of Triphenylmethanol (Grignard Reaction) Experiment Procedure (Printable pdf format) Introduction In this two-week experiment, triphenylmethanol will be synthesized through a Grignard reaction. Students will work individually to prepare the Grignard reagent by reacting bromobenzene with solid magnesium in diethylether. The prepared Grignard reagent, phenylmagnesium bromide, will then be combined with bezophenone to form the desired triphenylmethanol product. Procedure NOTE: All glassware must be extremely dry! Place glassware in the drying oven for at least 10 minutes before beginning the reaction. Preparation of the Grignard Reagent Add ~0.5g of magnesium turnings and a magnetic stir bar to a dry 100 ml round-bottomed flask, and place the flask in the drying oven for 30 minutes. Remove the flask from the oven, clamp it to a ring stand, insert a reflux condenser, fit the flask into a heating mantle and immediately attach a drying tube to the top of the reflux condenser. Position the flask in the heating mantle on a stirrer/hotplate. Set the reaction up on the side of the hood near to the faucet labeled for distillation. Allow the reaction flask to cool to room temperature completely before proceeding. While the flask is cooling, place a 150ml beaker into the drying oven. While the flask is cooling, add 3.5 g of dry bromobenzene to a dry 50 mL Erlenmeyer flask and dissolve it in 5.0 mL of anhydrous diethyl ether. Use a glass powder funnel to dispense the bromobenze and ether to the Erlenmyer flask. -
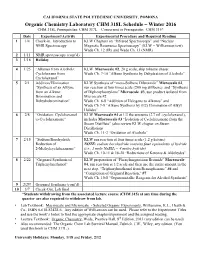
318 Lab Schedule
CALIFORNIA STATE POLYTECHNIC UNIVERSITY, POMONA Organic Chemistry Laboratory CHM 318L Schedule – Winter 2016 CHM 318L Prerequisites: CHM 317L Concurrent or Prerequisite: CHM 315* Date Experiment/Activity Experimental Procedure and Required Reading 1 1/4 Check-in. Introduction to KLW Chapters on “Infrared Spectroscopy” and “Nuclear NMR Spectroscopy Magnetic Resonance Spectroscopy” (KLW = Williamson text) Wade Ch. 12 (IR) and Wade Ch. 13 (NMR) 2 1/11 NMR spectroscopy (cont’d) 3 1/18 Holiday 4 1/25 “Alkenes from Alcohols: KLW Macroscale #2, 20 g scale, skip toluene chaser Cyclohexene from Wade Ch. 7-10 “Alkene Synthesis by Dehydration of Alcohols” Cyclohexanol” 5 2/1 Addition/Elimination KLW Synthesis of “meso-Stilbene Dibromide” Microscale #2, “Synthesis of an Alkyne run reaction at four times scale (200 mg stillbene); and “Synthesis from an Alkene: of Diphenylacetylene” Microscale #3, use product isolated from Bromination and Microscale #2 Dehydrobromination” Wade Ch. 8-8 “Addition of Halogens to Alkenes” and Wade Ch 7-9 “Alkene Synthesis by (E2) Elimination of Alkyl Halides” 6 2/8 “Oxidation: Cyclohexanol KLW Macroscale #4 at 1/3 the amounts (2.7 mL cyclohexanol), to Cyclohexanone” includes Macroscale #3 “Isolation of Cyclohexanone from the Steam Distillate” (also review KLW chapter on Steam Distillation) Wade Ch. 11-2 “Oxidation of Alcohols” 7 2/15 “Sodium Borohydride KLW run reaction at four times scale (1.2 g ketone) Reduction of NOTE: sodium borohydride contains four equivalents of hydride 2-Methylcyclohexanone” (i.e., 1 mole NaBH4 = 4 moles hydride) Wade Ch. 10-11 & 18-20 “Reductions of Ketones & Aldehydes” 8 2/22 “Grignard Synthesis of KLW preparation of “Phenylmagnesium Bromide” Macroscale Triphenylmethanol” #4, run reaction at 1/2 scale and then use the entire amount in the next step, “Triphenylmethanol from Benzophenone” #6 and “Completion of Grignard Reaction,” #7 Wade Ch. -

Revised Group Additivity Values for Enthalpies of Formation (At 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds
Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon– Hydrogen and Carbon–Hydrogen–Oxygen Compounds Cite as: Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 Submitted: 17 January 1996 . Published Online: 15 October 2009 N. Cohen ARTICLES YOU MAY BE INTERESTED IN Additivity Rules for the Estimation of Molecular Properties. Thermodynamic Properties The Journal of Chemical Physics 29, 546 (1958); https://doi.org/10.1063/1.1744539 Critical Evaluation of Thermochemical Properties of C1–C4 Species: Updated Group- Contributions to Estimate Thermochemical Properties Journal of Physical and Chemical Reference Data 44, 013101 (2015); https:// doi.org/10.1063/1.4902535 Estimation of the Thermodynamic Properties of Hydrocarbons at 298.15 K Journal of Physical and Chemical Reference Data 17, 1637 (1988); https:// doi.org/10.1063/1.555814 Journal of Physical and Chemical Reference Data 25, 1411 (1996); https://doi.org/10.1063/1.555988 25, 1411 © 1996 American Institute of Physics for the National Institute of Standards and Technology. Revised Group Additivity Values for Enthalpies of Formation (at 298 K) of Carbon-Hydrogen and Carbon-Hydrogen-Oxygen Compounds N. Cohen Thermochemical Kinetics Research, 6507 SE 31st Avenue, Portland, Oregon 97202-8627 Received January 17, 1996; revised manuscript received September 4, 1996 A program has been undertaken for the evaluation and revision of group additivity values (GAVs) necessary for predicting, by means of Benson's group additivity method, thermochemical properties of organic molecules. This review reports on the portion of that program dealing with GAVs for enthalpies of formation at 298.15 K (hereinafter abbreviated as 298 K) for carbon-hydrogen and carbon-hydrogen-oxygen compounds. -

Grignard Synthesis of Triphenylmethanol Reactions That Form Carbon-Carbon Bonds Are Among the Most Useful to the Synthetic Organic Chemist
1 Experiment 12: Grignard Synthesis of Triphenylmethanol Reactions that form carbon-carbon bonds are among the most useful to the synthetic organic chemist. In 1912, Victor Grignard received the Nobel prize in chemistry for his discovery of a new series of reactions that result in the formation of a carbon-carbon bond. A Grignard synthesis first involves the preparation of an organomagnesium reagent via the reaction of an alkyl bromide with magnesium metal: δ– δ+ R Br + Mg R MgBr The resulting “Grignard reagent” acts as both a good nucleophile and a strong base. Its nucleophilic character allows it to react with the electrophilic carbon in a carbonyl group, thus forming the carbon-carbon bond. Its basic property means that it will react with acidic compounds, such as carboxylic acids, phenols, thiols and even alcohols and water; therefore, reaction conditions must be free from acids and strictly anhydrous. Grignard reagents will also react with oxygen to form hydroperoxides, thus they are highly unstable when exposed to the atmosphere and are generally not isolated from solution. For a variety of reasons, anhydrous diethyl ether is the solvent of choice for carrying out a Grignard synthesis. Vapors from the highly volatile solvent help to prevent oxygen from reaching the reaction solution. In addition, evidence suggests that the ether molecules actually coordinate with and help stabilize the Grignard reagent: Et Et O R Mg Br O Et Et The magnesium metal used in the synthesis contains a layer of oxide on the surface that prevents it from reacting with the alkyl bromide. The pieces of metal must be gently scratched while in the ether solution to expose fresh surface area so that the reaction can commence. -

The Effects of Stoichiometry and Starting Material on the Product Identity and Yield in Grignard Addition Reactions
Supplementary information for Comprehensive Organic Chemistry Experiments for the Laboratory Classroom © The Royal Society of Chemistry 2017 The effects of stoichiometry and starting material on the product identity and yield in Grignard addition reactions Supplementary Material This experiment has been performed both in the 150 person standard introductory organic chemistry laboratory (taught primarily by undergraduate teaching assistants in five sections of 30-40 students) and in a special introductory organic chemistry laboratory for freshman, taught to 23 students in sections of 7 and 16. This course is the first organic laboratory for these students but has been taught in the spring semester along with the second semester of organic lecture. The lab periods for this course are five hours long, and this experiment is typically performed in the latter half of the semester. This experiment is used to illustrate to students the importance of planning their time in lab; they must be out of the lab in the 5 hours allotted. While many students finish the experiment in this time, others plan to finish the following week along with a shorter experiment. Those who were running behind were encouraged to finish through the drying of their organic layer with MgSO4 and set up the distillation the following week. When doing this they should make sure that their organic layer is in a closed container to prevent evaporation of their product. Moisture Sensitive Conditions There are many diverse protocols for maintaining the anhydrous conditions required to successfully prepare and utilize Grignard reagents. We typically open a fresh can of anhydrous ether and dispense it directly, without additional drying in a still or air sensitive techniques for solvent transfers. -

CHM205 Chemicals by Experiment Tuesday, November 17, 2015 3:14:15 PM Experiment Title Chemical Name Concentration Acetaminophen Synthesis Acetic Anhydride Liquid
CHM205 Chemicals by Experiment Tuesday, November 17, 2015 3:14:15 PM Experiment Title Chemical Name Concentration Acetaminophen Synthesis Acetic anhydride liquid Acetaminophen Synthesis p-aminophenol solid Alcohols to Alkyl chlorides 2-pentanol liquid Alcohols to Alkyl chlorides Hydrochloric acid 12 M Alcohols to Alkyl chlorides Sodium carbonate solid Alcohols to Alkyl chlorides Hydrobromic acid 48% w/v Alcohols to Alkyl chlorides Sodium sulfate anhydrous solid Alcohols to Alkyl chlorides sec-phenethyl alcohol liquid Alcohols to Alkyl chlorides Benzyl alcohol liquid Alcohols to Alkyl chlorides t-butanol liquid Alcohols to Alkyl chlorides 1-pentanol liquid Alcohols to Alkyl chlorides Sodium carbonate 10% w/v Diels Alder Reaction 2,3-dimethyl-1,3-butadiene liquid Diels Alder Reaction Maleic anhydride solid Diels Alder Reaction Ethanol 95% Liquid Diels Alder Reaction Hexane liquid Diels Alder Reaction Cyclohexane liquid Diels Alder Reaction Calcium chloride solid Esterification methanol liquid Esterification Sodium carbonate 10% w/v Esterification 1-propanol liquid Esterification 1-butanol liquid Esterification trans-cinnamic acid solid Esterification Isoamyl alcohol liquid Esterification Isopropyl alcohol liquid Esterification Benzyl alcohol liquid Esterification Sulfuric acid conc. 18 M Esterification 1-pentanol liquid Esterification Isobutyl alcohol liquid Esterification Ethanol 95% liquid Page 1 of 3 Experiment Title Chemical Name Concentration Extraction of Beta Carotene Cyclohexane liquid Extraction of Beta Carotene Beta carotene UV -

Organic Chemistry Laboratory Experiments for Organic Chemistry Laboratory 860-121-02 Mw 1:00-4:00
ORGANIC CHEMISTRY LABORATORY EXPERIMENTS FOR ORGANIC CHEMISTRY LABORATORY 860-121-02 MW 1:00-4:00 WRITTEN, COMPILED AND EDITED BY LINDA PAAR JEFFREY ELBERT KIRK MANFREDI SPRING 2008 TABLE OF CONTENTS SYNTHESIS OF ASPIRIN 1 MELTING POINT AND CRYSTALLIZATION 2 DISTILLATION 8 EXTRACTION 11 TLC AND CHROMATOGRAPHY 14 NATURAL PRODUCTS: ISOLATION OF LIMONENE 23 FREE RADICAL CHLORINATION 24 SN1 AND SN2 REACTIONS 27 DEHYDRATION REACTIONS 30 GRIGNARD SYNTHESIS 32 COMPUTATIONAL CHEMISTRY 36 MULTIPLE STEP SYNTHESIS 38 ORGANIC CHEMISTRY 121 EXPERIMENT 1 SYNTHESIS OF ASPIRIN FROM SALICYLIC ACID Aspirin is one of the oldest and most common drugs in use today. It is both an analgesic (pain killer) and antipyretic (reduces fever). One method of preparation is to react salicylic acid (1 ) with acetic anhydride (2) and a trace amount of acid (equation 1). O OH CH3 O COOH COOH + H + CH3COOH + (CH3CO)2O 3 4 1 2 The chemical name for aspirin is acetylsalicylic acid (3) PROCEDURE Place 3.00 g of salicylic acid in a 125 ml Erlenmeyer flask. Cautiously add 6 ml of acetic anhydride and then 5 drops of concentrated H2SO4. Mix the reagents and heat the flask in a beaker of water warmed to 80-90°C, for 10 minutes. Remove the Erlenmeyer flask and allow it to cool to room temperature. Add 40 ml of H2O and let the sample crystallize in an ice-water bath.* Filter and wash the crystals with cold water. Allow them to air dry overnight and weigh the product. What is the percent yield? One drawback to this synthetic procedure is that there is the possibility of some left over salicylic acid. -

Organic Chemistry, 5E (Bruice) Chapter 17: Carbonyl Compounds II
Organic Chemistry, 5e (Bruice) Chapter 17: Carbonyl Compounds II 1) Which of the following compounds is an aldehyde? A) I B) II C) III D) IV E) V Answer: D Type: MC Section: 17-1 2) Which of the following compounds is a ketone? A) I B) II C) III D) IV E) V Answer: B Type: MC Section: 17-1 3) What is the common name for the following compound? A) chloroaldehyde B) α-chloroacetaldehyde C) β-chloroacetaldehyde D) 2-chloroethanal E) α-chloroethanal Answer: B Type: MC Section: 17-1 4) Which of the following compounds is acetone? A) B) C) D) E) Answer: A Type: MC Section: 17-1 5) What is the IUPAC name for the following compound? A) 2-bromobutanal B) α-bromobutanal C) 3-bromobutanal D) β-bromobutyraldehyde E) 3-bromobutanone Answer: C Type: MC Section: 17-1 6) Which of the following compounds is benzaldehyde? A) I B) II C) III D) IV E) V Answer: D Type: MC Section: 17-1 7) What is the IUPAC name for the following compound? A) 5-chloro-3-methylhexanone B) 1-chloro-1,3-dimethyl-4-pentanone C) 5-chloro-3,5-dimethyl-2-pentanone D) 5-chloro-3-methyl-2-hexanone E) 2-chloro-4-methyl-5-hexanone Answer: D Type: MC Section: 17-1 8) Which of the following is cyclohexanone? A) I B) II C) III D) IV E) V Answer: E Type: MC Section: 17-1 9) Provide the proper IUPAC name for PhCH 2CH(CH 3)CH 2CH 2CHO. Answer: 4-methyl-5-phenylpentanal Type: SA Section: 17-1 10) Provide the proper IUPAC name for CH 3CHOHCH 2COCH 2C(CH 3)2CH 2CH 3. -

Ezp 36 Triphenylmethanol and Benzoic Acid-2
Triphenylmethanol and Benzoic Acid Grignard reaction Extraction Crystallization In this experiment, you will prepare a Grignard reagent, or organomagnesium reagent. The reagent is phenylmagnesium bromide. ether Bromobenzene Phenylmagnesium Bromide This reagent will be converted to a tertiary alcohol or a carboxylic acid, depending on the experiment selected. O + ether - + H30 C C Benzophenone C Triphenylmethanol 1 O - + + ether H3O C O C OH MgBr(OH) Benzoic Acid The alkyl portion of the Grignard reagent behaves as if it had the characteristics of a carbanion. We may write the structure of the reagent as a partially ionic compound: δ- δ+ R MgX This partially bonded carbanion is a Lewis base. It reacts with strong acids, as you would expect, to give an alkane: δ- δ+ R MgX + HX → R — H + MgX2 Any compound with a suitably acidic hydrogen will donate a proton to destroy the reagent. Water, alcohols, terminal acetylenes, phenols, and carboxylic acids are all acidic enough to bring about this reaction. The Grignard reagent also functions as a good nucleophile in nucleophilic addition reactions of the carbonyl group. The carbonyl group has electrophilic character at its carbon atom (due to resonance), and a good nucleophile seeks out this center for addition. .. - ::O : O : δ+ .. δ- C O C C .. + 2 The magnesium salts produced form a complex with the addition product, an alkoxide salt. In a second step of the reaction, these must be hydrolyzed (protonated) by addition of dilute aqueous acid: O O MgX OH HX C + RMgX C C MgX2 H2O + R R Step 1 Step 2 The Grignard reaction is used synthetically to prepare secondary alcohols from aldehydes and tertiary alcohols from ketones. -

Alcohols and Ethers
ALCOHOLS AND ETHERS The hydroxyl group is one of the most important functional groups of na- turally occurring organic molecules. All carbohydrates and their derivatives, including nucleic acids, have hydroxyl groups. Some amino acids, most ste- roids, many terpenes, and plant pigments have hydroxyl groups. These sub- stances serve many diverse purposes for the support and maintenance of life. One extreme example is the potent toxin tetrodotoxin, which is isolated from puffer fish and has obvious use for defense against predators. This compound has special biochemical interest, having six different hydroxylic functions arranged on a cagelike structure: tetrodotoxin On the more practical side, vast quantities of simple alcohols - metha- nol, ethanol, 2-propanol, 1-butan01 - and many ethers are made from petro- leum-derived hydrocarbons. These alcohols are widely used as solvents and as intermediates for the synthesis of more complex substances. 15 Alcohols and Ethers The reactions involving the hydrogens of alcoholic OH groups are expected to be similar to those of water, HOH, the simplest hydroxylic com- pound. Alcohols, ROH, can be regarded in this respect as substitution products of water. However, with alcohols we shall be interested not only in reactions that proceed at the 8-H bond but also with processes that result in cleavage of the C-0 bond, or changes in the organic group R. The simple ethers, ROR, do not have 0-H bonds, and most of their reactions are limited to the substituent groups. The chemistry of ethers, there- fore, is less varied than that of alcohols. This fact is turned to advantage in the widespread use of ethers as solvents for a variety of organic reactions, as we already have seen for Grignard reagents (Section 14- 10).