Alcohols and Ethers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 21 the Chemistry of Carboxylic Acid Derivatives
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 21 The Chemistry of Carboxylic Acid Derivatives Solutions to In-Text Problems 21.1 (b) (d) (e) (h) 21.2 (a) butanenitrile (common: butyronitrile) (c) isopentyl 3-methylbutanoate (common: isoamyl isovalerate) The isoamyl group is the same as an isopentyl or 3-methylbutyl group: (d) N,N-dimethylbenzamide 21.3 The E and Z conformations of N-acetylproline: 21.5 As shown by the data above the problem, a carboxylic acid has a higher boiling point than an ester because it can both donate and accept hydrogen bonds within its liquid state; hydrogen bonding does not occur in the ester. Consequently, pentanoic acid (valeric acid) has a higher boiling point than methyl butanoate. Here are the actual data: INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 21 2 21.7 (a) The carbonyl absorption of the ester occurs at higher frequency, and only the carboxylic acid has the characteristic strong, broad O—H stretching absorption in 2400–3600 cm–1 region. (d) In N-methylpropanamide, the N-methyl group is a doublet at about d 3. N-Ethylacetamide has no doublet resonances. In N-methylpropanamide, the a-protons are a quartet near d 2.5. In N-ethylacetamide, the a- protons are a singlet at d 2. The NMR spectrum of N-methylpropanamide has no singlets. 21.9 (a) The first ester is more basic because its conjugate acid is stabilized not only by resonance interaction with the ester oxygen, but also by resonance interaction with the double bond; that is, the conjugate acid of the first ester has one more important resonance structure than the conjugate acid of the second. -

The Progression of Chiral Anions from Concepts to Applications in Asymmetric Catalysis Robert J
REVIEW ARTICLE PUBLISHED ONLINE: 24 JULY 2012 | DOI: 10.1038/NCHEM.1405 The progression of chiral anions from concepts to applications in asymmetric catalysis Robert J. Phipps, Gregory L. Hamilton and F. Dean Toste* Despite the tremendous advances of the past four decades, chemists are far from being able to use chiral catalysts to con- trol the stereoselectivity of any desired reaction. New concepts for the construction and mode of operation of chiral catalysts have the potential to open up previously inaccessible reaction space. The recognition and categorization of distinct approaches seems to play a role in triggering rapid exploration of new territory. This Review both reflects on the origins as well as details a selection of the latest examples of an area that has advanced considerably within the past five years or so: the use of chiral anions in asymmetric catalysis. Defining reactions as involving chiral anions is a difficult task owing to uncertainties over the exact catalytic mechanisms. Nevertheless, we attempt to provide an overview of the breadth of reactions that could reasonably fall under this umbrella. 9 ithin the broader field of science, organic chemistry is Of significantly greater acidity (pKa around 2–4) are chiral phos- often referred to as a mature field of study. In spite of this, phoric acids, commonly derived from BINOL (1,1′-bi-2-naphthol). Wnew avenues of inquiry seem to arise with relative fre- These catalysts have risen to prominence over the past decade quency. Perhaps we take it for granted that expansions of the field because their high acidity permits activation of a broad range of are sparked when one or two chemical laboratories uncover a new substrates3,10. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

United States Patent 15) 3,669,956 Borck Et Al
United States Patent 15) 3,669,956 Borck et al. (45) June 13, 1972 54) 4-SUBSTITUTEDAMNO 260/472, 260/516, 260/518 R, 260/518 A, 260/519, PHENYACETIC ACDS AND 260/556 AR, 260/556 B, 260/558 S, 260/558 A, DERVATIVES THEREOF 260/559 T, 260/559 A, 260/.571, 260/574, 260/.575, (72 Inventors: Joachim Borck; Johann Dahin; Volker 424/244, 424/246, 424/248, 424/250, 424/267, Koppe; Josef Kramer; Gustav Shorre; J. 424/270, 424/272, 424/273, 424/274, 424/304, W. Hermann Hovy; Ernst Schorscher, all 424/309, 424/32 i, 424/324, 424/330 of Darmstadt, Germany 51) int. Cl. ........................................................ C07d 41/04 58) Field of Search........ 260/294X,293.4, 293.47, 239 BF, 73) Assignee: E. Merck A. G., Darmstadt, Germany 260/326.3, 294.3 E (22) Filed: July 22, 1968 56) References Cited (21) Appl. No.: 746,326 UNITED STATES PATENTS (30) Foreign Application Priority Data 3,252,970 5/1966 Huebner................................ 260/239 July 22, 1967 Germany.............................. M 74881 3,385,852 5/1968 Casadio................................. 260/246 Jan. 8, 1968 Germany... ....M 76850 OTHER PUBLICATIONS Feb. 23, 1968 Germany...... ...M 77363 March 1, 1968 Germany.............................. M 77429 Norman et al., J. Chen. Soc. 1963, (Nov.), 5431-6. (52) U.S. Cl................... 260/239 BF, 260/239 A, 260/239 E, Primary Examiner-Henry R. Jiles 260/243 B, 260/246, 260/247. 1, 260/247.2 R, Assistant Examiner-G. Thomas Todd 260/247.2 A, 260/247.2 B, 260/247.5 R, 260/247.7 Attorney-Millen, Raptes & White A, 260/247.7 H, -

Exam 1 (February 23, 2004) ID# ______
Chemistry 211 Name ___________________________ Exam 1 (February 23, 2004) ID# ___________________________ 10 1. You desire to synthesize 3-ethyl-3-pentanol starting with an ester. (i) What would be the name of the ester, and what is the name for the Grignard reagent (e.g., methyl magnesium bromide)? (ii) For the carbons shown in the product, show plausible hydrocarbons that you could start with to produce the ester and the Grignard reagent (as in a retrosynthesis). 12 2. (i) Show the step-by-step process required to produce propyllithium, which requires a free radical reaction mechanism, . (ii) Show the complete reaction mechanism for reaction between propyllithium and the correct ketone to produce 3-propyl-3-pentanol. (iii) Propose a possible reaction mechanism by which dipropyl cuprate (Cu+ with two propyl groups attached) could react with ethyl bromide to produce a new hydrocarbon. (This is a thinking exercise! So, think! () 8 3. As mentioned in the text, diethyl ether, pentane, and 1-butanol have similar molar masses, but different physical properties. Boiling points are 35oC, 36oC, and 117oC, respectively. Their respective solubilities in water are 7.5g/100mL, insoluble, and 9g/100mL. (i) Draw structures for each of these compounds. (ii) Justify the observed boiling points and their solubilities. 16 4. Draw structures of the following compounds 2,3-heptanediol isopropyllithium benzylmagnesium bromide benzoic acid benzaldehylde dimethyl sulfide t-butyl methanoate dibutyl ketone 12 5. Alcohols can be oxidized to produce other compounds, and can be produced by reduction. For the reactions shown below, show the structure for the expected product (if reaction does not occur, state: No Reaction) when treated with the indicated oxidizing or reducing agents. -
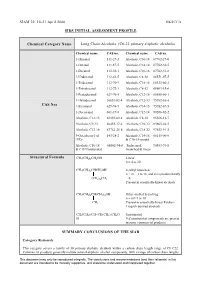
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

Grignard Reaction: Synthesis of Triphenylmethanol
*NOTE: Grignard reactions are very moisture sensitive, so all the glassware in the reaction (excluding the work-up) should be dried in an oven with a temperature of > 100oC overnight. The following items require oven drying. They should be placed in a 150mL beaker, all labeled with a permanent marker. 1. 5mL conical vial (AKA: Distillation receiver). 2. Magnetic spin vane. 3. Claisen head. 4. Three Pasteur pipettes. 5. Two 1-dram vials (Caps EXCLUDED). 6. One 2-dram vial (Caps EXCLUDED). 7. Glass stirring rod 8. Adaptor (19/22.14/20) Grignard Reaction: Synthesis of Triphenylmethanol Pre-Lab: In the “equations” section, besides the main equations, also: 1) draw the equation for the production of the byproduct, Biphenyl. 2) what other byproduct might occur in the reaction? Why? In the “observation” section, draw data tables in the corresponding places, each with 2 columns -- one for “prediction” (by answering the following questions) and one for actual drops or observation. 1) How many drops of bromobenzene should you add? 2) How many drops of ether will you add to flask 2? 3) 100 µL is approximately how many drops? 4) What are the four signs of a chemical reaction? (Think back to Chem. 110) 5) How do the signs of a chemical reaction apply to this lab? The Grignard reaction is a useful synthetic procedure for forming new carbon- carbon bonds. This organometallic chemical reaction involves alkyl- or aryl-magnesium halides, known as Grignard 1 reagents. Grignard reagents are formed via the action of an alkyl or aryl halide on magnesium metal. -

Physicochemical Properties of Organic Medicinal Agents
Principles of Drug Action 1, Spring 2005, Esters ESTERS AND RELATED CARBOXYLIC ACID DERIVATIVES Jack DeRuiter I. Structure and Preparation Esters are derivatives of carboxylic acids that arise via replacement of the hydroxyl (OH) portion of the acid COOH function with an "ether" moiety (-OR): O O H C C O C O Acid Ester Note that replacement of the acid OH group with an "ether" moiety removes the acidic function from the parent structure (acid) resulting in the formation of non-acidic (neutral, but somewhat polar) compounds (esters). Esters can be sub-classified based on their general structure as aliphatic, aromatic or cyclic (called "lactones") as illustrated by the examples below: O O CH2CH3 CH2CH3 O CH3 O O O Aliphatic Ester Aromatic Ester Cyclic Ester (Lactone) A variety of methods have been developed for the preparation of esters. Most of these methods involve reaction of an alcohol with an "activated carboxylic acid" compound (i.e. acid chloride): O O H C C X OC C O X- Ester "Activated" acid (X=Cl) Alcohol (Electrophile) (Nucleophile) The ester functionality does not introduce a center of asymmetry and thus optical and geometric isomerism does not result from the presence of this functional group. The ester functionality (the carbonyl and ether oxygen) is composed of an sp2 hybridized carbon so it cannot be chiral, and since there is free rotation about the ether bond geometric isomerism also is not possible at the sp2 center. 1 Principles of Drug Action 1, Spring 2005, Esters II. Solubility of Esters Esters contain carbonyl (C=O) and ether (O-C) dipoles arising from covalent bonding between electronegative oxygen atoms and electronically neutral carbon atoms. -

Halogenated Ether, Alcohol, and Alkane Anesthetics Activate TASK-3 Tandem Pore Potassium Channels Likely Through a Common Mechanism S
Supplemental material to this article can be found at: http://molpharm.aspetjournals.org/content/suppl/2017/03/21/mol.117.108290.DC1 1521-0111/91/6/620–629$25.00 https://doi.org/10.1124/mol.117.108290 MOLECULAR PHARMACOLOGY Mol Pharmacol 91:620–629, June 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics Halogenated Ether, Alcohol, and Alkane Anesthetics Activate TASK-3 Tandem Pore Potassium Channels Likely through a Common Mechanism s Anita Luethy, James D. Boghosian, Rithu Srikantha, and Joseph F. Cotten Department of Anesthesia, Critical Care, and Pain Medicine, Massachusetts General Hospital, Boston, Massachusetts (A.L., J.D.B., and J.F.C.); Department of Anesthesia, Kantonsspital Aarau, Aarau, Switzerland (A.L.); Carver College of Medicine, University of Iowa, Iowa City, Iowa (R.S.) Received January 7, 2017; accepted March 20, 2017 Downloaded from ABSTRACT The TWIK-related acid-sensitive potassium channel 3 (TASK-3; hydrate (165% [161–176]) . 2,2-dichloro- . 2-chloro 2,2,2- KCNK9) tandem pore potassium channel function is activated by trifluoroethanol . ethanol. Similarly, carbon tetrabromide (296% halogenated anesthetics through binding at a putative anesthetic- [245–346]), carbon tetrachloride (180% [163–196]), and 1,1,1,3,3,3- binding cavity. To understand the pharmacologic requirements for hexafluoropropanol (200% [194–206]) activate TASK-3, whereas molpharm.aspetjournals.org TASK-3 activation, we studied the concentration–response of the larger carbon tetraiodide and a-chloralose inhibit. Clinical TASK-3 to several anesthetics (isoflurane, desflurane, sevoflurane, agents activate TASK-3 with the following rank order efficacy: halothane, a-chloralose, 2,2,2-trichloroethanol [TCE], and chloral halothane (207% [202–212]) . -

Lecture 10 Carbon-Nitrogen Bonds Formation I
NPTEL – Chemistry – Principles of Organic Synthesis Lecture 10 Carbon-Nitrogen Bonds Formation I 4.1 Principles The methods for the formation of bonds between nitrogen and aliphatic carbon can be broadly divided into two categories: (i) reaction of nucleophilic nitrogen with electrophilic carbon, and (ii) reaction of electrophilic nitrogen with nucleophilic carbon. In this section, we will try to cover some of the important reactions. 4.2 Substitution of Nitrogen Nucleophiles at Saturated Carbon 4.2.1 Ritter Reaction Treatment of a tertiary alcohol or the corresponding alkene with concentrated sulphuric acid and a nitrile affords an amide that in acidic conditions undergoes hydrolysis to give amine. First, a R H, RCN H3O + RCO H OH N O NH 2 H2O H 2 Mechanism OH2 H -H2O :NC-R H2O OH OH N N R N R 2 C R OH O H2O proton H3O + RCO2H N R N R NH2 -H3O transfer H Scheme 1 tertiary carbocation is formed which is attacked by the nitrile to give a quaternary ion. The latter is decomposed by water to provide an amide which, in acidic conditions, is hydrolyzed to give an amine (Scheme 1). Joint initiative of IITs and IISc – Funded by MHRD Page 1 of 35 NPTEL – Chemistry – Principles of Organic Synthesis Examples: Me H N H2SO4 + N Chlorobenzene O 91% S. J. Chang, Organic Process Research and Development 1999, 3, 232. Me Me Me Me Me Me Me Me SnCl4 MeCN CH2 CH2 Cl N N T.-L. Ho, R.-J. Chein, J. Org. Chem. 2004, 69, 591. Me Me O Ph HNCHO Ph OH Ph HN H2SO4 Me H2SO4 N TMSCN N MeCN N Bn Bn Bn H. -

Chip Incompatibility Filters
ChIP Incompatibility Filters Filter Name Type Description includes carboxylic acid halides and >1 acyl halide and related SMARTS derivatives like chloroformates, carbamoyl- , imidoyl halides, etc. >1 aldehyde SMARTS R no heteroatom no isocyanate, ketene, etc. >1 alkyl bromide / iodide SMARTS no acyl halide or related or vinyl halide >1 amine aromatic primary SMARTS aromatic carbon bound to N, N not charged >1 amines (aromatic/aliphatic, primary no amide, enamine, etc., no heteroatom SMARTS or secondary) bound to N, N not charged no amide, enamine, etc, no heteroatom >1 amines nucleophilic (aliphatic SMARTS bound to N, no aromatic carbon bound to primary or secondary) N, N not charged >1 aryl bromide / iodide SMARTS any aryl bromide / iodide >1 aryl halide SMARTS any aryl halide any boronic acid derivative, aromatic or >1 boronic acid derivative SMARTS aliphatic >1 carbonyl acid SMARTS any carboxylic or carbamic acid, etc. >1 carboxylic acid anhydrides SMARTS carbon must be bound to carbonyl no heteroatom bound to carbonyl or >1 carboxylic acid ester SMARTS oxygen, no acid, no anydride, etc >1 isocyanate / isothiocyanate SMARTS no restrictions to nitrogen substituents R no heteroatom, no isocyanate, ketene, >1 ketone or aldehyde SMARTS etc. >1 NH any SMARTS R can be anything >1 thioamide and related (any) SMARTS any substitution >1 thiol and related (nucleophic) SMARTS any SH or negative S >2 NH any SMARTS R can be anything acidic compounds I combination sulfonyl acids and carboxylic acids anhydrides, bicarbonates, thio and imino acyl anhydrides and derivatives SMARTS derivatives, etc. includes carboxylic acid halides and acyl halide and related SMARTS derivatives like chloroformates, carbamoyl- , imidoyl halides, etc.