Kegggraph: a Graph Approach to KEGG PATHWAY in R and Bioconductor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Kyoto Encyclopedia of Genes and Genomes (KEGG)
Kyoto Encyclopedia of Genes and Genome Minoru Kanehisa Institute for Chemical Research, Kyoto University HFSPO Workshop, Strasbourg, November 18, 2016 The KEGG Databases Category Database Content PATHWAY KEGG pathway maps Systems information BRITE BRITE functional hierarchies MODULE KEGG modules KO (KEGG ORTHOLOGY) KO groups for functional orthologs Genomic information GENOME KEGG organisms, viruses and addendum GENES / SSDB Genes and proteins / sequence similarity COMPOUND Chemical compounds GLYCAN Glycans Chemical information REACTION / RCLASS Reactions / reaction classes ENZYME Enzyme nomenclature DISEASE Human diseases DRUG / DGROUP Drugs / drug groups Health information ENVIRON Health-related substances (KEGG MEDICUS) JAPIC Japanese drug labels DailyMed FDA drug labels 12 manually curated original DBs 3 DBs taken from outside sources and given original annotations (GENOME, GENES, ENZYME) 1 computationally generated DB (SSDB) 2 outside DBs (JAPIC, DailyMed) KEGG is widely used for functional interpretation and practical application of genome sequences and other high-throughput data KO PATHWAY GENOME BRITE DISEASE GENES MODULE DRUG Genome Molecular High-level Practical Metagenome functions functions applications Transcriptome etc. Metabolome Glycome etc. COMPOUND GLYCAN REACTION Funding Annual budget Period Funding source (USD) 1995-2010 Supported by 10+ grants from Ministry of Education, >2 M Japan Society for Promotion of Science (JSPS) and Japan Science and Technology Agency (JST) 2011-2013 Supported by National Bioscience Database Center 0.8 M (NBDC) of JST 2014-2016 Supported by NBDC 0.5 M 2017- ? 1995 KEGG website made freely available 1997 KEGG FTP site made freely available 2011 Plea to support KEGG KEGG FTP academic subscription introduced 1998 First commercial licensing Contingency Plan 1999 Pathway Solutions Inc. -

Precise Generation of Systems Biology Models from KEGG Pathways Clemens Wrzodek*,Finjabuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell
Wrzodek et al. BMC Systems Biology 2013, 7:15 http://www.biomedcentral.com/1752-0509/7/15 METHODOLOGY ARTICLE OpenAccess Precise generation of systems biology models from KEGG pathways Clemens Wrzodek*,FinjaBuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell Abstract Background: The KEGG PATHWAY database provides a plethora of pathways for a diversity of organisms. All pathway components are directly linked to other KEGG databases, such as KEGG COMPOUND or KEGG REACTION. Therefore, the pathways can be extended with an enormous amount of information and provide a foundation for initial structural modeling approaches. As a drawback, KGML-formatted KEGG pathways are primarily designed for visualization purposes and often omit important details for the sake of a clear arrangement of its entries. Thus, a direct conversion into systems biology models would produce incomplete and erroneous models. Results: Here, we present a precise method for processing and converting KEGG pathways into initial metabolic and signaling models encoded in the standardized community pathway formats SBML (Levels 2 and 3) and BioPAX (Levels 2 and 3). This method involves correcting invalid or incomplete KGML content, creating complete and valid stoichiometric reactions, translating relations to signaling models and augmenting the pathway content with various information, such as cross-references to Entrez Gene, OMIM, UniProt ChEBI, and many more. Finally, we compare several existing conversion tools for KEGG pathways and show that the conversion from KEGG to BioPAX does not involve a loss of information, whilst lossless translations to SBML can only be performed using SBML Level 3, including its recently proposed qualitative models and groups extension packages. -
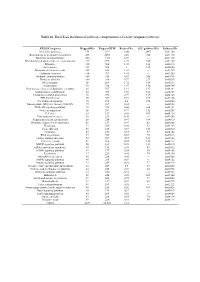
Table 1S. the KEGG Biochemical Pathways Categorization of LA Lily Unigenes Pathways
Table 1S. The KEGG biochemical pathways categorization of LA lily unigenes pathways. KEGG Categories Mapped-KO Unigene-NUM Ratio of No. ALL pathway KO Pathway-ID Metabolic pathways 954 4337 8.66 2067 ko01100 Biosynthesis of secondary metabolites 403 2285 4.56 720 ko01110 Biosynthesis of antibiotics 206 1147 2.29 --- ko01130 Microbial metabolism in diverse environments 157 995 1.99 720 ko01120 Ribosome 122 504 1.01 142 ko03010 Spliceosome 107 502 1 115 ko03040 Biosynthesis of amino acids 105 614 1.23 --- ko01230 Carbon metabolism 104 707 1.41 --- ko01200 Oxidative phosphorylation 100 335 0.67 206 ko00190 Purine metabolism 100 386 0.77 237 ko00230 RNA transport 98 861 1.72 134 ko03013 Endocytosis 92 736 1.47 138 ko04144 Protein processing in endoplasmic reticulum 88 567 1.13 137 ko04141 homologous recombination 84 933 1.86 144 ko05169 Ubiquitin mediated proteolysis 78 396 0.79 119 ko04120 HTLV-I infection 76 367 0.73 199 ko05166 Pyrimidine metabolism 76 298 0.6 150 ko00240 Non-alcoholic fatty liver disease (NAFLD) 72 209 0.42 --- ko04932 PI3K-Akt signaling pathway 71 328 0.66 226 ko04151 Viral carcinogenesis 68 387 0.77 132 ko05203 Cell cycle 63 339 0.68 103 ko04110 Proteoglycans in cancer 58 229 0.46 --- ko05205 Regulation of actin cytoskeleton 58 234 0.47 144 ko04810 Ribosome biogenesis in eukaryotes 56 237 0.47 82 ko03008 Phagosome 54 280 0.56 93 ko04145 Focal adhesion 53 185 0.37 133 ko04510 Lysosome 53 253 0.51 99 ko04142 RNA degradation 53 305 0.61 70 ko03018 Herpes simplex infection 52 257 0.51 121 ko05168 Cell cycle - yeast 51 260 -

A Tool for Retrieving Pathway Genes from KEGG Pathway Database
bioRxiv preprint doi: https://doi.org/10.1101/416131; this version posted September 13, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. KPGminer: A tool for retrieving pathway genes from KEGG pathway database A. K. M. Azad School of Biotechnology and Biomolecular Sciences, University of NSW, Chancellery Walk, Kensington, 2033, Australia Abstract Pathway analysis is a very important aspect in computational systems bi- ology as it serves as a crucial component in many computational pipelines. KEGG is one of the prominent databases that host pathway information associated with various organisms. In any pathway analysis pipelines, it is also important to collect and organize the pathway constituent genes for which a tool to automatically retrieve that would be a useful one to the practitioners. In this article, I present KPGminer, a tool that retrieves the constituent genes in KEGG pathways for various organisms and or- ganizes that information suitable for many downstream pathway analysis pipelines. We exploited several KEGG web services using REST APIs, par- ticularly GET and LIST methods to request for the information retrieval which is available for developers. Moreover, KPGminer can operate both for a particular pathway (single mode) or multiple pathways (batch mode). Next, we designed a crawler to extract necessary information from the re- sponse and generated outputs accordingly. KPGminer brings several key features including organism-specific and pathway-specific extraction of path- way genes from KEGG and always up-to-date information. -

Downregulated Developmental Processes in the Postnatal Right
www.nature.com/cddiscovery ARTICLE OPEN Downregulated developmental processes in the postnatal right ventricle under the influence of a volume overload ✉ ✉ ✉ Chunxia Zhou1,6, Sijuan Sun2,6, Mengyu Hu3, Yingying Xiao1, Xiafeng Yu1 , Lincai Ye 1,4,5 and Lisheng Qiu 1 © The Author(s) 2021 The molecular atlas of postnatal mouse ventricular development has been made available and cardiac regeneration is documented to be a downregulated process. The right ventricle (RV) differs from the left ventricle. How volume overload (VO), a common pathologic state in children with congenital heart disease, affects the downregulated processes of the RV is currently unclear. We created a fistula between the abdominal aorta and inferior vena cava on postnatal day 7 (P7) using a mouse model to induce a prepubertal RV VO. RNAseq analysis of RV (from postnatal day 14 to 21) demonstrated that angiogenesis was the most enriched gene ontology (GO) term in both the sham and VO groups. Regulation of the mitotic cell cycle was the second-most enriched GO term in the VO group but it was not in the list of enriched GO terms in the sham group. In addition, the number of Ki67-positive cardiomyocytes increased approximately 20-fold in the VO group compared to the sham group. The intensity of the vascular endothelial cells also changed dramatically over time in both groups. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the downregulated transcriptome revealed that the peroxisome proliferators-activated receptor (PPAR) signaling pathway was replaced by the cell cycle in the top-20 enriched KEGG terms because of the VO. -

Transcriptome Analysis of the Regulatory Mechanism of Foxo on Wing Dimorphism in the Brown Planthopper, Nilaparvata Lugens (Hemiptera: Delphacidae)
insects Article Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae) Nan Xu 1, Sheng-Fei Wei 1 and Hai-Jun Xu 1,2,3,* 1 State Key Laboratory of Rice Biology, Zhejiang University, Hangzhou 310058, China; [email protected] (N.X.); [email protected] (S.-F.W.) 2 Ministry of Agriculture Key Laboratory of Molecular Biology of Crop Pathogens and Insect Pests, Zhejiang University, Hangzhou 310058, China 3 Institute of Insect Sciences, Zhejiang University, Hangzhou 310058, China * Correspondence: [email protected]; Tel.: +86-571-88982996 Simple Summary: The brown planthopper (BPH) Nilaparvata lugens can develop into either long- winged or short-winged adults depending on environmental stimuli received during larval stages. The transcription factor NlFoxO serves as a key regulator determining alternative wing morphs in BPH, but the underlying molecular mechanism is largely unknown. Here, we investigated the transcriptomic profile of forewing and hindwing buds across the 5th-instar stage, the wing-morph decision stage. Our results indicated that NlFoxO modulated the developmental plasticity of wing buds mainly by regulating the expression of cell proliferation-associated genes. Abstract: The brown planthopper (BPH), Nilaparvata lugens, can develop into either short-winged (SW) or long-winged (LW) adults according to environmental conditions, and has long served as a model organism for exploring the mechanisms of wing polyphenism in insects. The transcription Citation: Xu, N.; Wei, S.-F.; Xu, H.-J. factor NlFoxO acts as a master regulator that directs the development of either SW or LW morphs, Transcriptome Analysis of the but the underlying molecular mechanism is largely unknown. -

"Using the KEGG Database Resource". In: Current Protocols in Bioinformatics
Using the KEGG Database Resource UNIT 1.12 KEGG (Kyoto Encyclopedia of Genes and Genomes) is a bioinformatics resource for understanding biological function from a genomic perspective. It is a multispecies, integrated resource consisting of genomic, chemical, and network information with cross-references to numerous outside databases and containing a complete set of building blocks (genes and molecules) and wiring diagrams (biological pathways) to represent cellular functions (Kanehisa et al., 2004). In this unit, protocols are described for using the five major KEGG resources: PATHWAY, GENES, SSDB, EXPRESSION, and LIGAND. The KEGG PATHWAY database (see Basic Protocols 1 to 4) consists of a user-friendly tool for analyzing the network of protein and small-molecule interactions that occur in the cells of various organisms. KEGG GENES (see Basic Protocols 5 and 6) provides access to the collection of gene data organized so as to be accessible via text searches, from the PATHWAY database, or via cross-species orthology searches. The KEGG Sequence Similiarity Database (SSDB; see Basic Protocols 7 to 9) consists of a precomputed database of all-versus-all Smith- Waterman similarity scores among all genes in KEGG GENES, enabling relationships between homologs to be easily visualized on the pathway and genome maps or viewed as clusters of orthologous genes. The KEGG EXPRESSION database (see Basic Protocols 10 to 14) contains both data and tools for analyzing gene expression data. User-defined data such as microarray experiments may also be uploaded for analysis using the tools available in KEGG EXPRESSION. Finally, KEGG LIGAND (see Basic Protocols 16 to 19) is a database of small molecules, their structures, and information relating to the enzymes that act on them. -

KEGG: Kyoto Encyclopedia of Genes and Genomes Hiroyuki Ogata, Susumu Goto, Kazushige Sato, Wataru Fujibuchi, Hidemasa Bono and Minoru Kanehisa*
1999 Oxford University Press Nucleic Acids Research, 1999, Vol. 27, No. 1 29–34 KEGG: Kyoto Encyclopedia of Genes and Genomes Hiroyuki Ogata, Susumu Goto, Kazushige Sato, Wataru Fujibuchi, Hidemasa Bono and Minoru Kanehisa* Institute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan Received September 8, 1998; Revised September 22, 1998; Accepted October 14, 1998 ABSTRACT The basic concepts of KEGG (1) and underlying informatics technologies (2,3) have already been published. KEGG is tightly Kyoto Encyclopedia of Genes and Genomes (KEGG) is integrated with the LIGAND chemical database for enzyme a knowledge base for systematic analysis of gene reactions (4,5) as well as with most of the major molecular functions in terms of the networks of genes and biology databases by the DBGET/LinkDB system (6) under the molecules. The major component of KEGG is the Japanese GenomeNet service (7). The database organization PATHWAY database that consists of graphical dia- efforts require extensive analyses of completely sequenced grams of biochemical pathways including most of the genomes, as exemplified by the analyses of metabolic pathways known metabolic pathways and some of the known (8) and ABC transport systems (9). In this article, we describe the regulatory pathways. The pathway information is also current status of the KEGG databases and discuss the use of represented by the ortholog group tables summarizing KEGG for functional genomics. orthologous and paralogous gene groups among different organisms. KEGG maintains the GENES OBJECTIVES OF KEGG database for the gene catalogs of all organisms with complete genomes and selected organisms with In May 1995, we initiated the KEGG project under the Human partial genomes, which are continuously re-annotated, Genome Program of the Ministry of Education, Science, Sports as well as the LIGAND database for chemical com- and Culture in Japan. -

RNA Seq Analyses of Chicken Reveals Biological Pathways Involved in Acclimation Into Diferent Geographical Locations Himansu Kumar1, Hyojun Choo2, Asankadyr U
www.nature.com/scientificreports OPEN RNA seq analyses of chicken reveals biological pathways involved in acclimation into diferent geographical locations Himansu Kumar1, Hyojun Choo2, Asankadyr U. Iskender1,3, Krishnamoorthy Srikanth1, Hana Kim1, Asankadyr T. Zhunushov3, Gul Won Jang1, Youngjo Lim1, Ki‑Duk Song4 & Jong‑Eun Park1* Transcriptome expression refects genetic response in diverse conditions. In this study, RNA sequencing was utilized to profle multiple tissues such as liver, breast, caecum, and gizzard of Korean commercial chicken raised in Korea and Kyrgyzstan. We analyzed ten samples per tissue from each location to identify candidate genes which are involved in the adaptation of Korean commercial chicken to Kyrgyzstan. At false discovery rate (FDR) < 0.05 and fold change (FC) > 2, we found 315, 196, 167 and 198 genes in liver, breast, cecum, and gizzard respectively as diferentially expressed between the two locations. GO enrichment analysis showed that these genes were highly enriched for cellular and metabolic processes, catalytic activity, and biological regulations. Similarly, KEGG pathways analysis indicated metabolic, PPAR signaling, FoxO, glycolysis/gluconeogenesis, biosynthesis, MAPK signaling, CAMs, citrate cycles pathways were diferentially enriched. Enriched genes like TSKU, VTG1, SGK, CDK2 etc. in these pathways might be involved in acclimation of organisms into diverse climatic conditions. The qRT‑PCR result also corroborated the RNA‑Seq fndings with R2 of 0.76, 0.80, 0.81, and 0.93 for liver, breast, caecum, and gizzard respectively. Our fndings can improve the understanding of environmental acclimation process in chicken. Recently, consumption of chicken meat has increased worldwide because of its rich nutrition, low cost, high quality protein, low cholesterol, and low fat1. -

GC-MS Metabolomics Reveals Metabolic Differences of the Farmed
www.nature.com/scientificreports OPEN GC-MS metabolomics reveals metabolic diferences of the farmed Mandarin fsh Siniperca chuatsi in recirculating ponds aquaculture system and pond Mingsong Xiao 1*, Kelin Qian2, Yuliang Wang1 & Fangyin Bao1 Siniperca chuatsi is currently one of the most important economic farmed freshwater fsh in China. The aim of this study was to evaluate the metabolic profle of recirculating ponds aquaculture system (RAS)- farmed S. chuatsi. Gas Chromatography-Mass Spectrophotometry (GC-MS) metabolomic platform was used to comprehensively analyze the efects of recirculating ponds aquaculture system (RAS) on the Mandarin fsh S. chuatsi metabolism. Database searching and statistical analysis revealed that there were altogether 335 metabolites quantifed (similarity > 0) and 205 metabolites were identifed by mass spectrum matching with a spectral similarity > 700. Among the 335 metabolites quantifed, 33 metabolites were signifcantly diferent (VIP > 1 and p < 0.05) between RAS and pond groups. In these thirty-three metabolites, taurine, 1-Hexadecanol, Shikimic Acid, Alloxanoic Acid and Acetaminophen were higher in the pond group, while 28 metabolites were increased notably in the RAS group. The biosynthesis of unsaturated fatty acids, lysosome, tryptophan metabolism were recommended as the KEGG pathway maps for S. chuatsi farmed in RAS. RAS can provide comprehensive benefts to the efects of Siniperca chuatsi metabolism, which suggest RAS is an efcient, economic, and environmentally friendly farming system compared to pond system. Te Chinese mandarin fsh Siniperca chuatsi (Basilewsky) is a freshwater fsh with high economic value and is endemic to East Asia, specially distributed in the Yangtze River drainage in China1. Te resources of wild S. -

Pathview: Pathway Based Data Integration and Visualization
Pathview: pathway based data integration and visualization Weijun Luo (luo weijun AT yahoo.com) May 19, 2021 Abstract In this vignette, we demonstrate the pathview package as a tool set for pathway based data integration and visualization. It maps and renders user data on relevant pathway graphs. All users need is to supply their gene or compound data and specify the target pathway. Pathview automatically downloads the pathway graph data, parses the data file, maps user data to the pathway, and renders pathway graph with the mapped data. Although built as a stand-alone program, pathview may seamlessly integrate with pathway and gene set (enrichment) analysis tools for a large-scale and fully automated analysis pipeline. In this vignette, we introduce common and advanced uses of pathview. We also cover package installation, data preparation, other useful features and common application errors. In gage package, vignette "RNA-Seq Data Pathway and Gene-set Analysis Workflows" demonstrates GAGE/Pathview workflows on RNA-seq (and microarray) pathway analysis. 1 Cite our work Please cite the Pathview paper formally if you use this package. This will help to support the maintenance and growth of the open source project. Weijun Luo and Cory Brouwer. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics, 29(14):1830-1831, 2013. doi: 10.1093/bioinformatics/btt285. 2 Quick start with demo data This is the most basic use of pathview, please check the full description below for details. Here we assume that the input data are already read in R as in the demo examples. -

FMAP: Functional Mapping and Analysis Pipeline for Metagenomics and Metatranscriptomics Studies Jiwoong Kim1,2†, Min Soo Kim1,2†, Andrew Y
Kim et al. BMC Bioinformatics (2016) 17:420 DOI 10.1186/s12859-016-1278-0 SOFTWARE Open Access FMAP: Functional Mapping and Analysis Pipeline for metagenomics and metatranscriptomics studies Jiwoong Kim1,2†, Min Soo Kim1,2†, Andrew Y. Koh2,4,5, Yang Xie1,2,3* and Xiaowei Zhan1,6* Abstract Background: Given the lack of a complete and comprehensive library of microbial reference genomes, determining the functional profile of diverse microbial communities is challenging. The available functional analysis pipelines lack several key features: (i) an integrated alignment tool, (ii) operon-level analysis, and (iii) the ability to process large datasets. Results: Here we introduce our open-sourced, stand-alone functional analysis pipeline for analyzing whole metagenomic and metatranscriptomic sequencing data, FMAP (Functional Mapping and Analysis Pipeline). FMAP performs alignment, gene family abundance calculations, and statistical analysis (three levels of analyses are provided: differentially-abundant genes, operons and pathways). The resulting output can be easily visualized with heatmaps and functional pathway diagrams. FMAP functional predictions are consistent with currently available functional analysis pipelines. Conclusion: FMAP is a comprehensive tool for providing functional analysis of metagenomic/metatranscriptomic sequencing data. With the added features of integrated alignment, operon-level analysis, and the ability to process large datasets, FMAP will be a valuable addition to the currently available functional analysis toolbox. We believe that this software will be of great value to the wider biology and bioinformatics communities. Background 16S rRNA sequences and gene content using whole Recent microbiome studies have revealed the complex genome shotgun (WGS) sequencing in order to identify the functional relationships between microorganisms and their functional capabilities of microbial communities.