A Decision Analysis Model for KEGG Pathway Analysis Junli Du1,2†, Manlin Li1†, Zhifa Yuan1, Mancai Guo1, Jiuzhou Song3, Xiaozhen Xie1 and Yulin Chen2*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Kyoto Encyclopedia of Genes and Genomes (KEGG)
Kyoto Encyclopedia of Genes and Genome Minoru Kanehisa Institute for Chemical Research, Kyoto University HFSPO Workshop, Strasbourg, November 18, 2016 The KEGG Databases Category Database Content PATHWAY KEGG pathway maps Systems information BRITE BRITE functional hierarchies MODULE KEGG modules KO (KEGG ORTHOLOGY) KO groups for functional orthologs Genomic information GENOME KEGG organisms, viruses and addendum GENES / SSDB Genes and proteins / sequence similarity COMPOUND Chemical compounds GLYCAN Glycans Chemical information REACTION / RCLASS Reactions / reaction classes ENZYME Enzyme nomenclature DISEASE Human diseases DRUG / DGROUP Drugs / drug groups Health information ENVIRON Health-related substances (KEGG MEDICUS) JAPIC Japanese drug labels DailyMed FDA drug labels 12 manually curated original DBs 3 DBs taken from outside sources and given original annotations (GENOME, GENES, ENZYME) 1 computationally generated DB (SSDB) 2 outside DBs (JAPIC, DailyMed) KEGG is widely used for functional interpretation and practical application of genome sequences and other high-throughput data KO PATHWAY GENOME BRITE DISEASE GENES MODULE DRUG Genome Molecular High-level Practical Metagenome functions functions applications Transcriptome etc. Metabolome Glycome etc. COMPOUND GLYCAN REACTION Funding Annual budget Period Funding source (USD) 1995-2010 Supported by 10+ grants from Ministry of Education, >2 M Japan Society for Promotion of Science (JSPS) and Japan Science and Technology Agency (JST) 2011-2013 Supported by National Bioscience Database Center 0.8 M (NBDC) of JST 2014-2016 Supported by NBDC 0.5 M 2017- ? 1995 KEGG website made freely available 1997 KEGG FTP site made freely available 2011 Plea to support KEGG KEGG FTP academic subscription introduced 1998 First commercial licensing Contingency Plan 1999 Pathway Solutions Inc. -

Precise Generation of Systems Biology Models from KEGG Pathways Clemens Wrzodek*,Finjabuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell
Wrzodek et al. BMC Systems Biology 2013, 7:15 http://www.biomedcentral.com/1752-0509/7/15 METHODOLOGY ARTICLE OpenAccess Precise generation of systems biology models from KEGG pathways Clemens Wrzodek*,FinjaBuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell Abstract Background: The KEGG PATHWAY database provides a plethora of pathways for a diversity of organisms. All pathway components are directly linked to other KEGG databases, such as KEGG COMPOUND or KEGG REACTION. Therefore, the pathways can be extended with an enormous amount of information and provide a foundation for initial structural modeling approaches. As a drawback, KGML-formatted KEGG pathways are primarily designed for visualization purposes and often omit important details for the sake of a clear arrangement of its entries. Thus, a direct conversion into systems biology models would produce incomplete and erroneous models. Results: Here, we present a precise method for processing and converting KEGG pathways into initial metabolic and signaling models encoded in the standardized community pathway formats SBML (Levels 2 and 3) and BioPAX (Levels 2 and 3). This method involves correcting invalid or incomplete KGML content, creating complete and valid stoichiometric reactions, translating relations to signaling models and augmenting the pathway content with various information, such as cross-references to Entrez Gene, OMIM, UniProt ChEBI, and many more. Finally, we compare several existing conversion tools for KEGG pathways and show that the conversion from KEGG to BioPAX does not involve a loss of information, whilst lossless translations to SBML can only be performed using SBML Level 3, including its recently proposed qualitative models and groups extension packages. -

COVID-19: Viral–Host Interactome Analyzed by Network Based
Messina et al. J Transl Med (2020) 18:233 https://doi.org/10.1186/s12967-020-02405-w Journal of Translational Medicine RESEARCH Open Access COVID-19: viral–host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection Francesco Messina1†, Emanuela Giombini1†, Chiara Agrati1, Francesco Vairo1, Tommaso Ascoli Bartoli1, Samir Al Moghazi1, Mauro Piacentini1,2, Franco Locatelli3, Gary Kobinger4, Markus Maeurer5,6, Alimuddin Zumla7,8, Maria R. Capobianchi1* , Francesco Nicola Lauria1†, Giuseppe Ippolito1† and COVID 19 INMI Network Medicine for IDs Study Group Abstract Background: Epidemiological, virological and pathogenetic characteristics of SARS-CoV-2 infection are under evaluation. A better understanding of the pathophysiology associated with COVID-19 is crucial to improve treatment modalities and to develop efective prevention strategies. Transcriptomic and proteomic data on the host response against SARS-CoV-2 still have anecdotic character; currently available data from other coronavirus infections are there- fore a key source of information. Methods: We investigated selected molecular aspects of three human coronavirus (HCoV) infections, namely SARS- CoV, MERS-CoV and HCoV-229E, through a network based-approach. A functional analysis of HCoV–host interactome was carried out in order to provide a theoretic host–pathogen interaction model for HCoV infections and in order to translate the results in prediction for SARS-CoV-2 pathogenesis. The 3D model of S-glycoprotein of SARS-CoV-2 was compared to the structure of the corresponding SARS-CoV, HCoV-229E and MERS-CoV S-glycoprotein. SARS-CoV, MERS-CoV, HCoV-229E and the host interactome were inferred through published protein–protein interactions (PPI) as well as gene co-expression, triggered by HCoV S-glycoprotein in host cells. -

Ingenuity Pathway Analysis of Human Facet Joint Tissues: Insight Into Facet Joint Osteoarthritis
EXPERIMENTAL AND THERAPEUTIC MEDICINE 19: 2997-3008, 2020 Ingenuity pathway analysis of human facet joint tissues: Insight into facet joint osteoarthritis CHU CHEN*, SHENGYU CUI*, WEIDONG LI, HURICHA JIN, JIANBO FAN, YUYU SUN and ZHIMING CUI Department of Spine Surgery, The Second Affiliated Hospital of Nantong University, Nantong, Jiangsu 226001, P.R. China Received August 17, 2019; Accepted January 30, 2020 DOI: 10.3892/etm.2020.8555 Abstract. Facet joint osteoarthritis (FJOA) is a common the top 5 IPA networks (with a score >30). The present study degenerative joint disorder with high prevalence in the elderly. provides insight into the pathological processes of FJOA from FJOA causes lower back pain and lower extremity pain, and a genetic perspective and may thus benefit the clinical treat- thus severely impacts the quality of life of affected patients. ment of FJOA. Emerging studies have focused on the histomorphological and histomorphometric changes in FJOA. However, the dynamic Introduction genetic changes in FJOA have remained to be clearly deter- mined. In the present study, previously obtained RNA deep Facet joint osteoarthritis (FJOA) is a common degenerative sequencing data were subjected to an ingenuity pathway joint disorder that causes the degeneration and breakdown analysis (IPA) and canonical signaling pathways of differ- of cartilage and restricts the movement of joints (1). It has entially expressed genes (DEGs) in FJOA were studied. The been reported that lumbar FJOA occurs at high prevalence top 25 enriched canonical signaling pathways were identified and the presence of FJOA is highly associated with age (2,3). and canonical signaling pathways with high absolute values A community-based cross-sectional study indicated that of z-scores, specifically leukocyte extravasation signaling, in populations aged <50 years, the prevalence of FJOA was Tec kinase signaling and osteoarthritis pathway, were inves- <45%, while it was ~75% in populations aged >50 years (4). -

Genomic-Wide Copy Number Variation Profile and a New IKZF1 Gene Variation in Chinese Adult Acute Lymphoblastic Leukemia
Research Article Clinics in Oncology Published: 02 Mar, 2021 Genomic-wide Copy Number Variation Profile and a New IKZF1 Gene Variation in Chinese Adult Acute Lymphoblastic Leukemia Tian Yuan1,2, Yingchan Wang1, Xiaoyan Li1, Yan Li1 and Yingchang Mi1* 1State Key Laboratory of Experimental Hematology, Chinese Academy of Medical Sciences and Peking Union Medical College, China 2Department of Hematology, Tianjin Medical University Cancer Institute and Hospital, China Abstract Purpose: Copy Number Variation (CNV) and Loss of Heterozygosity (LOH) were investigated in adult Chinese patients with Acute Lymphoblastic Leukemia (ALL), and these patients were screened for adult ALL prognostic genes. Methods: The CNV and LOH were detected with Affymetrix SNP 6.0 array. Using java and software R to construct the signal-net based on KEGG, enrichment the significant CNV genes associated with disease. Check the copy number variation of IKZF1 gene by FISH. Results: In Ph+ B-ALL patients, the most frequent CNVs were gain at chr13q, loss at chr6 and recurrent LOH at chr17p and 1p33. In Ph- B-ALL patients, the most frequent CNVs were gain at chr7, loss at chr9p and recurrent LOH at chr15q. In T-ALL, the most frequent CNVs were gain at chr20 and 7q, loss at chr2p and recurrent LOH also at chr15q. These CNV regions included several candidate genes such as IKZF1, JAK2, CDKN2A/2B, ETV6 etc. EGFR and IKBKG are in the center position of the Signal-Net, which may play essential role in ALL. The copy number amplification of IKZF1 gene is confirmed by FISH. OPEN ACCESS Discussion: CNV and LOH identification in adult ALL patients can identify susceptible genes, *Correspondence: guide diagnostic classification, and help with treatment selection. -

Recurrent Copy Number Variants Associated with Bronchopulmonary Dysplasia
nature publishing group Articles Translational Investigation Recurrent copy number variants associated with bronchopulmonary dysplasia Ausaf Ahmad1,2, Soumyaroop Bhattacharya1,2, Arthi Sridhar3, Anwar M. Iqbal3 and Thomas J. Mariani1,2 BACKGROUND: Variability in the incidence and severity of and almost all infants (>97%) with birth weight <1,250 g are bronchopulmonary dysplasia (BPD) among premature infants diagnosed with BPD (5). suggests that genetic susceptibility plays a role in pathogen- A diagnosis of BPD is dependent upon the use of supple- esis. An assessment of copy number variants (CNV) in BPD sub- mental oxygen following preterm birth. In addition to pre- jects may help to identify loci that harbor genetic susceptibility mature birth, environmental factors such as oxidative stress, factors. mechanical ventilation, and infection play a significant role METHODS: We conducted a retrospective analysis of clinical in pathogenesis of BPD. Higher than physiological levels of DNA microarray data from our institution. We identified 19 BPD therapeutic oxygen (hyperoxia) induce stress with production subjects, and 2 controls groups (full-term and preterm) with of reactive oxygen species. The lungs of premature infants are no lung-related disease. We reanalyzed raw data from each of susceptible to injury with pre- and postnatal exposures. These these subjects to identify recurrent CNV loci in BPD subjects. exposures may cause lung damage and induce a deviation from RESULTS: We identified three loci (at 11q13.2, 16p13.3, and the normal developmental path (4). 22q11.23–q12.1) with recurrent CNV in BPD subjects. The fre- Variability in the incidence and severity of BPD among pre- quency of these CNV was significantly higher in BPD subjects mature infants with similar risk factors suggests that genetic when compared with at least one control group. -
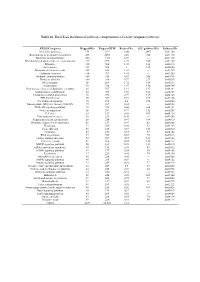
Table 1S. the KEGG Biochemical Pathways Categorization of LA Lily Unigenes Pathways
Table 1S. The KEGG biochemical pathways categorization of LA lily unigenes pathways. KEGG Categories Mapped-KO Unigene-NUM Ratio of No. ALL pathway KO Pathway-ID Metabolic pathways 954 4337 8.66 2067 ko01100 Biosynthesis of secondary metabolites 403 2285 4.56 720 ko01110 Biosynthesis of antibiotics 206 1147 2.29 --- ko01130 Microbial metabolism in diverse environments 157 995 1.99 720 ko01120 Ribosome 122 504 1.01 142 ko03010 Spliceosome 107 502 1 115 ko03040 Biosynthesis of amino acids 105 614 1.23 --- ko01230 Carbon metabolism 104 707 1.41 --- ko01200 Oxidative phosphorylation 100 335 0.67 206 ko00190 Purine metabolism 100 386 0.77 237 ko00230 RNA transport 98 861 1.72 134 ko03013 Endocytosis 92 736 1.47 138 ko04144 Protein processing in endoplasmic reticulum 88 567 1.13 137 ko04141 homologous recombination 84 933 1.86 144 ko05169 Ubiquitin mediated proteolysis 78 396 0.79 119 ko04120 HTLV-I infection 76 367 0.73 199 ko05166 Pyrimidine metabolism 76 298 0.6 150 ko00240 Non-alcoholic fatty liver disease (NAFLD) 72 209 0.42 --- ko04932 PI3K-Akt signaling pathway 71 328 0.66 226 ko04151 Viral carcinogenesis 68 387 0.77 132 ko05203 Cell cycle 63 339 0.68 103 ko04110 Proteoglycans in cancer 58 229 0.46 --- ko05205 Regulation of actin cytoskeleton 58 234 0.47 144 ko04810 Ribosome biogenesis in eukaryotes 56 237 0.47 82 ko03008 Phagosome 54 280 0.56 93 ko04145 Focal adhesion 53 185 0.37 133 ko04510 Lysosome 53 253 0.51 99 ko04142 RNA degradation 53 305 0.61 70 ko03018 Herpes simplex infection 52 257 0.51 121 ko05168 Cell cycle - yeast 51 260 -

Gene Expression Profiling and Bioinformatic Analysis of Rabbit
ogy & N ol eu ur e ro N p h f y o s l i Matsuo, et al., J Neurol Neurophysiol 2014, 5:3 o a l n o r g u y o J Journal of Neurology & Neurophysiology DOI: 10.4172/2155-9562.1000201 ISSN: 2155-9562 Research Article Open Access Gene Expression Profiling and Bioinformatic Analysis of Rabbit Basilar Artery after Experimental Subarachnoid Hemorrhage Satoshi Matsuo, Yuichiro Kikkawa*, Masaaki Hokama, Ryota Kurogi, Akira Nakamizo, Masahiro Mizoguchi and Tomio Sasaki Department of Neurosurgery, Graduate School of Medical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan *Corresponding author: Yuichiro Kikkawa, Department of Neurosurgery, Graduate School of Medical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan, Tel: +81-92-642-5524; Fax: +81-92-642-5526; E-mail: [email protected] Received date: Feb 14, 2014, Accepted date: Mar 20, 2014, Published date: Mar 26, 2014 Copyright: © 2014 Matsuo S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Objective: The molecular mechanisms which contribute to the development of vascular events including cerebral vasospasm after subarachnoid hemorrhage (SAH) in cerebral artery remain to be elucidated. In this study, we investigated the time course of changes in the gene expression of cerebral artery using rabbit SAH model and performed bioinformatic analysis of differentially expressed genes. Methods: Rabbit basilar arteries were harvested on days 3, 5, and 7 after initial hemorrhage. -

A Pathway Analysis Method for Genome-Wide Association Studies Babak Shahbaba,A Catherine M
Research Article Received 22 March 2011, Accepted 2 November 2011 Published online in Wiley Online Library (wileyonlinelibrary.com) DOI: 10.1002/sim.4477 A pathway analysis method for genome-wide association studies Babak Shahbaba,a Catherine M. Shachafb and Zhaoxia Yua*† For genome-wide association studies, we propose a new method for identifying significant biological pathways. In this approach, we aggregate data across single-nucleotide polymorphisms to obtain summary measures at the gene level. We then use a hierarchical Bayesian model, which takes the gene-level summary measures as data, in order to evaluate the relevance of each pathway to an outcome of interest (e.g., disease status). Although shifting the focus of analysis from individual genes to pathways has proven to improve the statistical power and provide more robust results, such methods tend to eliminate a large number of genes whose pathways are unknown. For these genes, we propose to use a Bayesian multinomial logit model to predict the associated pathways by using the genes with known pathways as the training data. Our hierarchical Bayesian model takes the uncertainty regard- ing the pathway predictions into account while assessing the significance of pathways. We apply our method to two independent studies on type 2 diabetes and show that the overlap between the results from the two studies is statistically significant. We also evaluate our approach on the basis of simulated data. Copyright © 2012 John Wiley & Sons, Ltd. Keywords: hierarchical; pathway; genome-wide; uncertainty 1. Introduction With the development of high-throughput genotyping technologies, genome-wide association studies (GWAS) have been widely used for identifying common genetic variants that contribute to human com- plex traits. -

Pathway-Based Analysis of Genome-Wide Association Data
fphar-09-00158 March 9, 2018 Time: 17:6 # 1 ORIGINAL RESEARCH published: 13 March 2018 doi: 10.3389/fphar.2018.00158 Pathway-Based Analysis of Genome-Wide Association Data Identified SNPs in HMMR as Biomarker for Chemotherapy- Induced Neutropenia in Breast Cancer Patients Edited by: Rick Kittles, Behzad Bidadi1†, Duan Liu1, Krishna R. Kalari2, Matthias Rubner3, Alexander Hein3, Irell and Manella Graduate School Matthias W. Beckmann3, Brigitte Rack4, Wolfgang Janni5, Peter A. Fasching3, of Biological Sciences, United States Richard M. Weinshilboum1 and Liewei Wang1* Reviewed by: Ming Ta Michael Lee, 1 Division of Clinical Pharmacology, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Geisinger Health System, Rochester, MN, United States, 2 Division of Biomedical Statistics and Informatics, Department of Health Sciences Research, United States Mayo Clinic, Rochester, MN, United States, 3 Department of Gynecology and Obstetrics, University Hospital Erlangen, Ken Batai, Friedrich-Alexander-University Erlangen-Nuremberg, Erlangen, Germany, 4 Department of Gynecology and Obstetrics, University of Arizona, United States Ludwig-Maximilians-Universität München, Munich, Germany, 5 Department of Gynecology and Obstetrics, University Hospital Ulm, Ulm, Germany *Correspondence: Liewei Wang [email protected] Neutropenia secondary to chemotherapy in breast cancer patients can be life- † Present address: threatening and there are no biomarkers available to predict the risk of drug-induced Behzad Bidadi, Merck & Co., Inc., North Wales, PA, neutropenia in those patients. We previously performed a genome-wide association United States study (GWAS) for neutropenia events in women with breast cancer who were treated with 5-fluorouracil, epirubicin and cyclophosphamide and recruited to the SUCCESS- Specialty section: This article was submitted to A trial. -

A Tool for Retrieving Pathway Genes from KEGG Pathway Database
bioRxiv preprint doi: https://doi.org/10.1101/416131; this version posted September 13, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. KPGminer: A tool for retrieving pathway genes from KEGG pathway database A. K. M. Azad School of Biotechnology and Biomolecular Sciences, University of NSW, Chancellery Walk, Kensington, 2033, Australia Abstract Pathway analysis is a very important aspect in computational systems bi- ology as it serves as a crucial component in many computational pipelines. KEGG is one of the prominent databases that host pathway information associated with various organisms. In any pathway analysis pipelines, it is also important to collect and organize the pathway constituent genes for which a tool to automatically retrieve that would be a useful one to the practitioners. In this article, I present KPGminer, a tool that retrieves the constituent genes in KEGG pathways for various organisms and or- ganizes that information suitable for many downstream pathway analysis pipelines. We exploited several KEGG web services using REST APIs, par- ticularly GET and LIST methods to request for the information retrieval which is available for developers. Moreover, KPGminer can operate both for a particular pathway (single mode) or multiple pathways (batch mode). Next, we designed a crawler to extract necessary information from the re- sponse and generated outputs accordingly. KPGminer brings several key features including organism-specific and pathway-specific extraction of path- way genes from KEGG and always up-to-date information. -

Systematic Analysis of Copy Number Variation Associated with Congenital Diaphragmatic Hernia
Systematic analysis of copy number variation associated with congenital diaphragmatic hernia Qihui Zhua,1, Frances A. Highb,c,d,1, Chengsheng Zhanga,1, Eliza Cerveiraa, Meaghan K. Russellb, Mauro Longonib,d, Maliackal P. Joyb, Mallory Ryana, Adam Mil-homensa, Lauren Bellfya, Caroline M. Colettib, Pooja Bhayanib, Regis Hilab, Jay M. Wilsonc,d, Patricia K. Donahoeb,d,2,3, and Charles Leea,e,2,3 aThe Jackson Laboratory for Genomic Medicine, Farmington, CT 06032; bPediatric Surgical Research Laboratories, Massachusetts General Hospital, Boston, MA 02114; cDepartment of Surgery, Boston Children’s Hospital, Boston, MA 02115; dHarvard Medical School, Boston, MA 02115; and eDepartment of Life Sciences, Ewha Womans University, Seoul 03760, South Korea Contributed by Patricia K. Donahoe, April 4, 2018 (sent for review August 22, 2017; reviewed by Sally A. Camper and Deborah Krakow) Congenital diaphragmatic hernia (CDH), characterized by malfor- mouse models were also later found to harbor rare and predicted mation of the diaphragm and hypoplasia of the lungs, is one of the pathogenic mutations in patients with CDH (8, 10, 13). most common and severe birth defects, and is associated with high Numerous studies have implicated copy number variants morbidity and mortality rates. There is growing evidence demon- (CNVs) in human disease with associated phenotypes ranging strating that genetic factors contribute to CDH, although the patho- from cognitive disabilities to predispositions to obesity, cancer, and genesis remains largely elusive. Single-nucleotide polymorphisms otherdiseases(14–17). Both inherited and de novo CNVs play a have been studied in recent whole-exome sequencing efforts, causative role in CDH (18–22), and at least 10% of CDH cases are but larger copy number variants (CNVs) have not yet been studied estimated to be caused by genomic imbalances (21).