RNA Seq Analyses of Chicken Reveals Biological Pathways Involved in Acclimation Into Diferent Geographical Locations Himansu Kumar1, Hyojun Choo2, Asankadyr U
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Kyoto Encyclopedia of Genes and Genomes (KEGG)
Kyoto Encyclopedia of Genes and Genome Minoru Kanehisa Institute for Chemical Research, Kyoto University HFSPO Workshop, Strasbourg, November 18, 2016 The KEGG Databases Category Database Content PATHWAY KEGG pathway maps Systems information BRITE BRITE functional hierarchies MODULE KEGG modules KO (KEGG ORTHOLOGY) KO groups for functional orthologs Genomic information GENOME KEGG organisms, viruses and addendum GENES / SSDB Genes and proteins / sequence similarity COMPOUND Chemical compounds GLYCAN Glycans Chemical information REACTION / RCLASS Reactions / reaction classes ENZYME Enzyme nomenclature DISEASE Human diseases DRUG / DGROUP Drugs / drug groups Health information ENVIRON Health-related substances (KEGG MEDICUS) JAPIC Japanese drug labels DailyMed FDA drug labels 12 manually curated original DBs 3 DBs taken from outside sources and given original annotations (GENOME, GENES, ENZYME) 1 computationally generated DB (SSDB) 2 outside DBs (JAPIC, DailyMed) KEGG is widely used for functional interpretation and practical application of genome sequences and other high-throughput data KO PATHWAY GENOME BRITE DISEASE GENES MODULE DRUG Genome Molecular High-level Practical Metagenome functions functions applications Transcriptome etc. Metabolome Glycome etc. COMPOUND GLYCAN REACTION Funding Annual budget Period Funding source (USD) 1995-2010 Supported by 10+ grants from Ministry of Education, >2 M Japan Society for Promotion of Science (JSPS) and Japan Science and Technology Agency (JST) 2011-2013 Supported by National Bioscience Database Center 0.8 M (NBDC) of JST 2014-2016 Supported by NBDC 0.5 M 2017- ? 1995 KEGG website made freely available 1997 KEGG FTP site made freely available 2011 Plea to support KEGG KEGG FTP academic subscription introduced 1998 First commercial licensing Contingency Plan 1999 Pathway Solutions Inc. -

Precise Generation of Systems Biology Models from KEGG Pathways Clemens Wrzodek*,Finjabuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell
Wrzodek et al. BMC Systems Biology 2013, 7:15 http://www.biomedcentral.com/1752-0509/7/15 METHODOLOGY ARTICLE OpenAccess Precise generation of systems biology models from KEGG pathways Clemens Wrzodek*,FinjaBuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell Abstract Background: The KEGG PATHWAY database provides a plethora of pathways for a diversity of organisms. All pathway components are directly linked to other KEGG databases, such as KEGG COMPOUND or KEGG REACTION. Therefore, the pathways can be extended with an enormous amount of information and provide a foundation for initial structural modeling approaches. As a drawback, KGML-formatted KEGG pathways are primarily designed for visualization purposes and often omit important details for the sake of a clear arrangement of its entries. Thus, a direct conversion into systems biology models would produce incomplete and erroneous models. Results: Here, we present a precise method for processing and converting KEGG pathways into initial metabolic and signaling models encoded in the standardized community pathway formats SBML (Levels 2 and 3) and BioPAX (Levels 2 and 3). This method involves correcting invalid or incomplete KGML content, creating complete and valid stoichiometric reactions, translating relations to signaling models and augmenting the pathway content with various information, such as cross-references to Entrez Gene, OMIM, UniProt ChEBI, and many more. Finally, we compare several existing conversion tools for KEGG pathways and show that the conversion from KEGG to BioPAX does not involve a loss of information, whilst lossless translations to SBML can only be performed using SBML Level 3, including its recently proposed qualitative models and groups extension packages. -

Next-Generation Sequencing Reveals DGUOK Mutations in Adult Patients with Mitochondrial DNA Multiple Deletions
doi:10.1093/brain/aws258 Brain 2012: 135; 3404–3415 | 3404 BRAIN A JOURNAL OF NEUROLOGY Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions Dario Ronchi,1 Caterina Garone,2,3 Andreina Bordoni,1 Purificacion Gutierrez Rios,2 4,5,6,7 8 1 1 8 Sarah E. Calvo, Michela Ripolone, Michela Ranieri, Mafalda Rizzuti, Luisa Villa, Downloaded from Francesca Magri,1 Stefania Corti,1,9 Nereo Bresolin,1,9,10 Vamsi K. Mootha,4,5,6,7 Maurizio Moggio,8 Salvatore DiMauro,2 Giacomo P. Comi1,9 and Monica Sciacco8 1 Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, http://brain.oxfordjournals.org/ IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milan, Italy 2 Department of Neurology, Columbia University Medical Centre, New York, NY 10032, USA 3 Human Genetics Joint PhD Programme, University of Turin, 10125 Turin, Italy and University of Bologna, 40125 Bologna, Italy 4 Center for Human Genetic Research, Massachusetts General Hospital, Boston, MA 02114, USA 5 Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114, USA 6 Department of Systems Biology, Harvard Medical School, Boston, MA 02115, USA 7 Broad Metabolism Initiative, Seven Cambridge Center, Broad Institute of Harvard and MIT, Cambridge, MA 02142, USA 8 Neuromuscular Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Dino Ferrari Centre, University of Milan, 20122 Milan, Italy 9 Centre of Excellence on -

CD29 Identifies IFN-Γ–Producing Human CD8+ T Cells With
+ CD29 identifies IFN-γ–producing human CD8 T cells with an increased cytotoxic potential Benoît P. Nicoleta,b, Aurélie Guislaina,b, Floris P. J. van Alphenc, Raquel Gomez-Eerlandd, Ton N. M. Schumacherd, Maartje van den Biggelaarc,e, and Monika C. Wolkersa,b,1 aDepartment of Hematopoiesis, Sanquin Research, 1066 CX Amsterdam, The Netherlands; bLandsteiner Laboratory, Oncode Institute, Amsterdam University Medical Center, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; cDepartment of Research Facilities, Sanquin Research, 1066 CX Amsterdam, The Netherlands; dDivision of Molecular Oncology and Immunology, Oncode Institute, The Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands; and eDepartment of Molecular and Cellular Haemostasis, Sanquin Research, 1066 CX Amsterdam, The Netherlands Edited by Anjana Rao, La Jolla Institute for Allergy and Immunology, La Jolla, CA, and approved February 12, 2020 (received for review August 12, 2019) Cytotoxic CD8+ T cells can effectively kill target cells by producing therefore developed a protocol that allowed for efficient iso- cytokines, chemokines, and granzymes. Expression of these effector lation of RNA and protein from fluorescence-activated cell molecules is however highly divergent, and tools that identify and sorting (FACS)-sorted fixed T cells after intracellular cytokine + preselect CD8 T cells with a cytotoxic expression profile are lacking. staining. With this top-down approach, we performed an un- + Human CD8 T cells can be divided into IFN-γ– and IL-2–producing biased RNA-sequencing (RNA-seq) and mass spectrometry cells. Unbiased transcriptomics and proteomics analysis on cytokine- γ– – + + (MS) analyses on IFN- and IL-2 producing primary human producing fixed CD8 T cells revealed that IL-2 cells produce helper + + + CD8 Tcells. -

Ejhg2013112.Pdf
European Journal of Human Genetics (2014) 22, 184–191 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg ARTICLE Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene Johanna Uusimaa1,2,17, Julie Evans3,17, Conrad Smith3, Anna Butterworth4, Kate Craig4, Neil Ashley1, Chunyan Liao1, Janet Carver1, Alan Diot1, Lorna Macleod1, Iain Hargreaves5, Abdulrahman Al-Hussaini6, Eissa Faqeih6, Ali Asery6, Mohammed Al Balwi7, Wafaa Eyaid8, Areej Al-Sunaid8, Deirdre Kelly9, Indra van Mourik9, Sarah Ball10, Joanna Jarvis11, Arundhati Mulay12, Nedim Hadzic13, Marianne Samyn13, Alastair Baker13, Shamima Rahman14, Helen Stewart15, Andrew AM Morris16, Anneke Seller3, Carl Fratter3, Robert W Taylor4 and Joanna Poulton*,1 Mitochondrial DNA (mtDNA) depletion syndromes (MDS) are severe autosomal recessive disorders associated with decreased mtDNA copy number in clinically affected tissues. The hepatocerebral form (mtDNA depletion in liver and brain) has been associated with mutations in the POLG, PEO1 (Twinkle), DGUOK and MPV17 genes, the latter encoding a mitochondrial inner membrane protein of unknown function. The aims of this study were to clarify further the clinical, biochemical, cellular and molecular genetic features associated with MDS due to MPV17 gene mutations. We identified 12 pathogenic mutations in the MPV17 gene, of which 11 are novel, in 17 patients from 12 families. All patients manifested liver disease. Poor feeding, hypoglycaemia, raised serum lactate, hypotonia and faltering growth were common presenting features. mtDNA depletion in liver was demonstrated in all seven cases where liver tissue was available. Mosaic mtDNA depletion was found in primary fibroblasts by PicoGreen staining. -

Clinical and Molecular Characterization of Three Patients
Mahjoub et al. BMC Medical Genetics (2019) 20:167 https://doi.org/10.1186/s12881-019-0893-9 CASE REPORT Open Access Clinical and molecular characterization of three patients with Hepatocerebral form of mitochondrial DNA depletion syndrome: a case series Ghazale Mahjoub1, Parham Habibzadeh1,2, Hassan Dastsooz1,3, Malihe Mirzaei1, Arghavan Kavosi1, Laila Jamali1, Haniyeh Javanmardi2, Pegah Katibeh4, Mohammad Ali Faghihi1,5 and Seyed Alireza Dastgheib6* Abstract Background: Mitochondrial DNA depletion syndromes (MDS) are clinically and phenotypically heterogeneous disorders resulting from nuclear gene mutations. The affected individuals represent a notable reduction in mitochondrial DNA (mtDNA) content, which leads to malfunction of the components of the respiratory chain. MDS is classified according to the type of affected tissue; the most common type is hepatocerebral form, which is attributed to mutations in nuclear genes such as DGUOK and MPV17. These two genes encode mitochondrial proteins and play major roles in mtDNA synthesis. Case presentation: In this investigation patients in three families affected by hepatocerebral form of MDS who were initially diagnosed with tyrosinemia underwent full clinical evaluation. Furthermore, the causative mutations were identified using next generation sequencing and were subsequently validated using sanger sequencing. The effect of the mutations on the gene expression was also studied using real-time PCR. A pathogenic heterozygous frameshift deletion mutation in DGUOK gene was identified in parents of two affected patients (c.706–707 + 2 del: p.k236 fs) presenting with jaundice, impaired fetal growth, low-birth weight, and failure to thrive who died at the age of 3 and 6 months in family I. Moreover, a novel splice site mutation in MPV17 gene (c.461 + 1G > C) was identified in a patient with jaundice, muscle weakness, and failure to thrive who died due to hepatic failure at the age of 4 months. -

Blood-Derived Mitochondrial DNA Copy Number Is Associated with Gene Expression Across Multiple
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.17.209023; this version posted July 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Blood-derived mitochondrial DNA copy number is associated with gene expression across multiple 2 tissues and is predictive for incident neurodegenerative disease 3 4 Stephanie Y. Yang1, Christina A. Castellani1, Ryan J. Longchamps1, Vamsee K. Pillalamarri1, Brian 5 O’Rourke2, Eliseo Guallar3, Dan E. Arking1,2 6 7 8 1McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, 9 Baltimore, MD 10 2Division of Cardiology, Department of Medicine, Johns Hopkins University School of Medicine, 11 Baltimore, MD 12 3Departments of Epidemiology and Medicine, and Welch Center for Prevention, Epidemiology, and 13 Clinical Research, Johns Hopkins University Bloomberg School of Public Health, Baltimore, MD 14 15 Email addresses: 16 [email protected] 17 [email protected] 18 [email protected] 19 [email protected] 20 [email protected] 21 [email protected] 22 [email protected] 23 24 25 26 27 28 29 30 31 32 33 34 35 *Correspondence and address for reprints to: 36 Dan E. Arking, Ph.D. 37 Johns Hopkins University School of Medicine 38 733 N Broadway Ave 39 Miller Research Building Room 459 40 Baltimore, MD 21205 41 (410) 502-4867 (ph) 42 [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.17.209023; this version posted July 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Mitochondrial Medicine 2019: Washington DC Scientific and Clinical Meetings June 26-29, 2019 Hilton Alexandria Mark Center Alexandria, VA
Mitochondrial Medicine 2019: Washington DC Scientific and Clinical Meetings June 26-29, 2019 Hilton Alexandria Mark Center Alexandria, VA 2019 Course Chairs: Amel Kara, MD and Carla Koehler, PhD 2019 CME Chair: Bruce H. Cohen, MD Mitochondrial Medicine 2019: Washington DC Scientific/Clinical Program June 26-29, 2019 Course Description The United Mitochondrial Disease Foundation and PeerPoint Medical Education Institute have joined efforts to sponsor and organize a CME-accredited symposium. Mitochondrial diseases are more common than previously recognized and mitochondrial pathophysiology is now a recognized part of many disease processes, including heart disease, cancer, AIDS and diabetes. There have been significant advances in the molecular genetics, proteomics, epidemiology and clinical aspects of mitochondrial pathophysiology. This conference is directed toward the scientist and clinician interested in all aspects of mitochondrial science. The content of this educational program was determined by rigorous assessment of educational needs and includes surveys, program feedback, expert faculty assessment, literature review, medical practice, chart review and new medical knowledge. The format will include didactic lectures from invited experts intermixed with peer-reviewed platform presentations. There will be ample time for professional discussion both in and out of the meeting room, and peer-reviewed poster presentations will be given throughout the meeting. This will be a four-day scientific meeting aimed at those with scientific and -
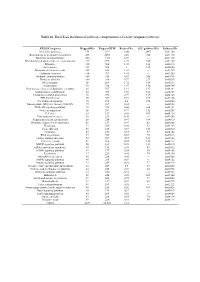
Table 1S. the KEGG Biochemical Pathways Categorization of LA Lily Unigenes Pathways
Table 1S. The KEGG biochemical pathways categorization of LA lily unigenes pathways. KEGG Categories Mapped-KO Unigene-NUM Ratio of No. ALL pathway KO Pathway-ID Metabolic pathways 954 4337 8.66 2067 ko01100 Biosynthesis of secondary metabolites 403 2285 4.56 720 ko01110 Biosynthesis of antibiotics 206 1147 2.29 --- ko01130 Microbial metabolism in diverse environments 157 995 1.99 720 ko01120 Ribosome 122 504 1.01 142 ko03010 Spliceosome 107 502 1 115 ko03040 Biosynthesis of amino acids 105 614 1.23 --- ko01230 Carbon metabolism 104 707 1.41 --- ko01200 Oxidative phosphorylation 100 335 0.67 206 ko00190 Purine metabolism 100 386 0.77 237 ko00230 RNA transport 98 861 1.72 134 ko03013 Endocytosis 92 736 1.47 138 ko04144 Protein processing in endoplasmic reticulum 88 567 1.13 137 ko04141 homologous recombination 84 933 1.86 144 ko05169 Ubiquitin mediated proteolysis 78 396 0.79 119 ko04120 HTLV-I infection 76 367 0.73 199 ko05166 Pyrimidine metabolism 76 298 0.6 150 ko00240 Non-alcoholic fatty liver disease (NAFLD) 72 209 0.42 --- ko04932 PI3K-Akt signaling pathway 71 328 0.66 226 ko04151 Viral carcinogenesis 68 387 0.77 132 ko05203 Cell cycle 63 339 0.68 103 ko04110 Proteoglycans in cancer 58 229 0.46 --- ko05205 Regulation of actin cytoskeleton 58 234 0.47 144 ko04810 Ribosome biogenesis in eukaryotes 56 237 0.47 82 ko03008 Phagosome 54 280 0.56 93 ko04145 Focal adhesion 53 185 0.37 133 ko04510 Lysosome 53 253 0.51 99 ko04142 RNA degradation 53 305 0.61 70 ko03018 Herpes simplex infection 52 257 0.51 121 ko05168 Cell cycle - yeast 51 260 -

A Tool for Retrieving Pathway Genes from KEGG Pathway Database
bioRxiv preprint doi: https://doi.org/10.1101/416131; this version posted September 13, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. KPGminer: A tool for retrieving pathway genes from KEGG pathway database A. K. M. Azad School of Biotechnology and Biomolecular Sciences, University of NSW, Chancellery Walk, Kensington, 2033, Australia Abstract Pathway analysis is a very important aspect in computational systems bi- ology as it serves as a crucial component in many computational pipelines. KEGG is one of the prominent databases that host pathway information associated with various organisms. In any pathway analysis pipelines, it is also important to collect and organize the pathway constituent genes for which a tool to automatically retrieve that would be a useful one to the practitioners. In this article, I present KPGminer, a tool that retrieves the constituent genes in KEGG pathways for various organisms and or- ganizes that information suitable for many downstream pathway analysis pipelines. We exploited several KEGG web services using REST APIs, par- ticularly GET and LIST methods to request for the information retrieval which is available for developers. Moreover, KPGminer can operate both for a particular pathway (single mode) or multiple pathways (batch mode). Next, we designed a crawler to extract necessary information from the re- sponse and generated outputs accordingly. KPGminer brings several key features including organism-specific and pathway-specific extraction of path- way genes from KEGG and always up-to-date information. -

Genetic Analysis of Mitochondrial DNA Copy Number and Associated Traits Identifies Loci Implicated In
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.25.428086; this version posted April 26, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Genetic analysis of mitochondrial DNA copy number and associated traits identifies loci implicated in 2 nucleotide metabolism, platelet activation, and megakaryocyte proliferation, and reveals a causal 3 association of mitochondrial function with mortality 4 Longchamps RJ1*, Yang SY1*, Castellani CA1,2, Shi W1, Lane J3, Grove ML4, Bartz 5 TM5, Sarnowski C6, Burrows K7,8, Guyatt AL9, Gaunt TR7,8, Kacprowski T10, Yang J11, De Jager PL12,13, Yu 6 L11, CHARGE Aging and Longevity Group, Bergman A14, Xia R15, Fornage M15,16, Feitosa 7 MF17, Wojczynski MK17, Kraja AT 17, Province MA 17, Amin N 18, Rivadeneira F19, Tiemeier H18,20, 8 Uitterlinden AG18,19, Broer L19, Van Meurs JBJ18,19, Van Duijn CM18, Raffield LM21, Lange L22, Rich 9 SS23, Lemaitre RN24, Goodarzi MO25, Sitlani CM24, Mak ACY26, Bennett DA11, Rodriguez S7,8, Murabito 10 JM27, Lunetta KL6, Sotoodehnia N28, Atzmon G29, Kenny Y30, Barzilai N31, Brody JA32, Psaty BM33, Taylor 11 KD34, Rotter JI34, Boerwinkle E4,35, Pankratz N3, Arking DE1 12 13 * Indicates authors contributed equally to this work. 14 1 McKusick-Nathans Institute, Department of Genetic Medicine, Johns Hopkins University School of 15 Medicine, Baltimore, MD 16 2 Department of Pathology -

Meta-Analysis of Gene Expression in Individuals with Autism Spectrum Disorders
Meta-analysis of Gene Expression in Individuals with Autism Spectrum Disorders by Carolyn Lin Wei Ch’ng BSc., University of Michigan Ann Arbor, 2011 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Bioinformatics) The University of British Columbia (Vancouver) August 2013 c Carolyn Lin Wei Ch’ng, 2013 Abstract Autism spectrum disorders (ASD) are clinically heterogeneous and biologically complex. State of the art genetics research has unveiled a large number of variants linked to ASD. But in general it remains unclear, what biological factors lead to changes in the brains of autistic individuals. We build on the premise that these heterogeneous genetic or genomic aberra- tions will converge towards a common impact downstream, which might be reflected in the transcriptomes of individuals with ASD. Similarly, a considerable number of transcriptome analyses have been performed in attempts to address this question, but their findings lack a clear consensus. As a result, each of these individual studies has not led to any significant advance in understanding the autistic phenotype as a whole. The goal of this research is to comprehensively re-evaluate these expression profiling studies by conducting a systematic meta-analysis. Here, we report a meta-analysis of over 1000 microarrays across twelve independent studies on expression changes in ASD compared to unaffected individuals, in blood and brain. We identified a number of genes that are consistently differentially expressed across studies of the brain, suggestive of effects on mitochondrial function. In blood, consistent changes were more difficult to identify, despite individual studies tending to exhibit larger effects than the brain studies.