Pathview: Pathway Based Data Integration and Visualization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Kyoto Encyclopedia of Genes and Genomes (KEGG)
Kyoto Encyclopedia of Genes and Genome Minoru Kanehisa Institute for Chemical Research, Kyoto University HFSPO Workshop, Strasbourg, November 18, 2016 The KEGG Databases Category Database Content PATHWAY KEGG pathway maps Systems information BRITE BRITE functional hierarchies MODULE KEGG modules KO (KEGG ORTHOLOGY) KO groups for functional orthologs Genomic information GENOME KEGG organisms, viruses and addendum GENES / SSDB Genes and proteins / sequence similarity COMPOUND Chemical compounds GLYCAN Glycans Chemical information REACTION / RCLASS Reactions / reaction classes ENZYME Enzyme nomenclature DISEASE Human diseases DRUG / DGROUP Drugs / drug groups Health information ENVIRON Health-related substances (KEGG MEDICUS) JAPIC Japanese drug labels DailyMed FDA drug labels 12 manually curated original DBs 3 DBs taken from outside sources and given original annotations (GENOME, GENES, ENZYME) 1 computationally generated DB (SSDB) 2 outside DBs (JAPIC, DailyMed) KEGG is widely used for functional interpretation and practical application of genome sequences and other high-throughput data KO PATHWAY GENOME BRITE DISEASE GENES MODULE DRUG Genome Molecular High-level Practical Metagenome functions functions applications Transcriptome etc. Metabolome Glycome etc. COMPOUND GLYCAN REACTION Funding Annual budget Period Funding source (USD) 1995-2010 Supported by 10+ grants from Ministry of Education, >2 M Japan Society for Promotion of Science (JSPS) and Japan Science and Technology Agency (JST) 2011-2013 Supported by National Bioscience Database Center 0.8 M (NBDC) of JST 2014-2016 Supported by NBDC 0.5 M 2017- ? 1995 KEGG website made freely available 1997 KEGG FTP site made freely available 2011 Plea to support KEGG KEGG FTP academic subscription introduced 1998 First commercial licensing Contingency Plan 1999 Pathway Solutions Inc. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Precise Generation of Systems Biology Models from KEGG Pathways Clemens Wrzodek*,Finjabuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell
Wrzodek et al. BMC Systems Biology 2013, 7:15 http://www.biomedcentral.com/1752-0509/7/15 METHODOLOGY ARTICLE OpenAccess Precise generation of systems biology models from KEGG pathways Clemens Wrzodek*,FinjaBuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell Abstract Background: The KEGG PATHWAY database provides a plethora of pathways for a diversity of organisms. All pathway components are directly linked to other KEGG databases, such as KEGG COMPOUND or KEGG REACTION. Therefore, the pathways can be extended with an enormous amount of information and provide a foundation for initial structural modeling approaches. As a drawback, KGML-formatted KEGG pathways are primarily designed for visualization purposes and often omit important details for the sake of a clear arrangement of its entries. Thus, a direct conversion into systems biology models would produce incomplete and erroneous models. Results: Here, we present a precise method for processing and converting KEGG pathways into initial metabolic and signaling models encoded in the standardized community pathway formats SBML (Levels 2 and 3) and BioPAX (Levels 2 and 3). This method involves correcting invalid or incomplete KGML content, creating complete and valid stoichiometric reactions, translating relations to signaling models and augmenting the pathway content with various information, such as cross-references to Entrez Gene, OMIM, UniProt ChEBI, and many more. Finally, we compare several existing conversion tools for KEGG pathways and show that the conversion from KEGG to BioPAX does not involve a loss of information, whilst lossless translations to SBML can only be performed using SBML Level 3, including its recently proposed qualitative models and groups extension packages. -

Oligosyndactylism Mice Have an Inversion of Chromosome 8
Copyright 2004 by the Genetics Society of America DOI: 10.1534/genetics.104.031914 Oligosyndactylism Mice Have an Inversion of Chromosome 8 Thomas L. Wise and Dimitrina D. Pravtcheva1 Department of Human Genetics, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, New York 10314 Manuscript received June 1, 2004 Accepted for publication August 4, 2004 ABSTRACT The radiation-induced mutation Oligosyndactylism (Os) is associated with limb and kidney defects in heterozygotes and with mitotic arrest and embryonic lethality in homozygotes. We reported that the cell cycle block in Os and in the 94-A/K transgene-induced mutations is due to disruption of the Anapc10 (Apc10/Doc1) gene. To understand the genetic basis of the limb and kidney abnormalities in Os mice we characterized the structural changes of chromosome 8 associated with this mutation. We demonstrate Mb) apart and the internal fragment 10ف) that the Os chromosome 8 has suffered two breaks that are 5 cM delineated by the breaks is in an inverted orientation on the mutant chromosome. While sequences in proximity to the distal break are present in an abnormal Os-specific Anapc10 hybrid transcript, transcrip- tion of these sequences in normal mice is low and difficult to detect. Transfer of the Os mutation onto an FVB/N background indicated that the absence of dominant effects in 94-A/K mice is not due to strain background effects on the mutation. Further analysis of this mutation will determine if a gene interrupted by the break or a long-range effect of the rearrangement on neighboring genes is responsible for the dominant effects of Os. -
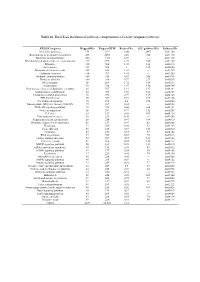
Table 1S. the KEGG Biochemical Pathways Categorization of LA Lily Unigenes Pathways
Table 1S. The KEGG biochemical pathways categorization of LA lily unigenes pathways. KEGG Categories Mapped-KO Unigene-NUM Ratio of No. ALL pathway KO Pathway-ID Metabolic pathways 954 4337 8.66 2067 ko01100 Biosynthesis of secondary metabolites 403 2285 4.56 720 ko01110 Biosynthesis of antibiotics 206 1147 2.29 --- ko01130 Microbial metabolism in diverse environments 157 995 1.99 720 ko01120 Ribosome 122 504 1.01 142 ko03010 Spliceosome 107 502 1 115 ko03040 Biosynthesis of amino acids 105 614 1.23 --- ko01230 Carbon metabolism 104 707 1.41 --- ko01200 Oxidative phosphorylation 100 335 0.67 206 ko00190 Purine metabolism 100 386 0.77 237 ko00230 RNA transport 98 861 1.72 134 ko03013 Endocytosis 92 736 1.47 138 ko04144 Protein processing in endoplasmic reticulum 88 567 1.13 137 ko04141 homologous recombination 84 933 1.86 144 ko05169 Ubiquitin mediated proteolysis 78 396 0.79 119 ko04120 HTLV-I infection 76 367 0.73 199 ko05166 Pyrimidine metabolism 76 298 0.6 150 ko00240 Non-alcoholic fatty liver disease (NAFLD) 72 209 0.42 --- ko04932 PI3K-Akt signaling pathway 71 328 0.66 226 ko04151 Viral carcinogenesis 68 387 0.77 132 ko05203 Cell cycle 63 339 0.68 103 ko04110 Proteoglycans in cancer 58 229 0.46 --- ko05205 Regulation of actin cytoskeleton 58 234 0.47 144 ko04810 Ribosome biogenesis in eukaryotes 56 237 0.47 82 ko03008 Phagosome 54 280 0.56 93 ko04145 Focal adhesion 53 185 0.37 133 ko04510 Lysosome 53 253 0.51 99 ko04142 RNA degradation 53 305 0.61 70 ko03018 Herpes simplex infection 52 257 0.51 121 ko05168 Cell cycle - yeast 51 260 -

Disruption of the Anaphase-Promoting Complex Confers Resistance to TTK Inhibitors in Triple-Negative Breast Cancer
Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer K. L. Thua,b, J. Silvestera,b, M. J. Elliotta,b, W. Ba-alawib,c, M. H. Duncana,b, A. C. Eliaa,b, A. S. Merb, P. Smirnovb,c, Z. Safikhanib, B. Haibe-Kainsb,c,d,e, T. W. Maka,b,c,1, and D. W. Cescona,b,f,1 aCampbell Family Institute for Breast Cancer Research, Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada M5G 1L7; bPrincess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada M5G 1L7; cDepartment of Medical Biophysics, University of Toronto, Toronto, ON, Canada M5G 1L7; dDepartment of Computer Science, University of Toronto, Toronto, ON, Canada M5G 1L7; eOntario Institute for Cancer Research, Toronto, ON, Canada M5G 0A3; and fDepartment of Medicine, University of Toronto, Toronto, ON, Canada M5G 1L7 Contributed by T. W. Mak, December 27, 2017 (sent for review November 9, 2017; reviewed by Mark E. Burkard and Sabine Elowe) TTK protein kinase (TTK), also known as Monopolar spindle 1 (MPS1), ator of the spindle assembly checkpoint (SAC), which delays is a key regulator of the spindle assembly checkpoint (SAC), which anaphase until all chromosomes are properly attached to the functions to maintain genomic integrity. TTK has emerged as a mitotic spindle, TTK has an integral role in maintaining genomic promising therapeutic target in human cancers, including triple- integrity (6). Because most cancer cells are aneuploid, they are negative breast cancer (TNBC). Several TTK inhibitors (TTKis) are heavily reliant on the SAC to adequately segregate their abnormal being evaluated in clinical trials, and an understanding of karyotypes during mitosis. -

A Tool for Retrieving Pathway Genes from KEGG Pathway Database
bioRxiv preprint doi: https://doi.org/10.1101/416131; this version posted September 13, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. KPGminer: A tool for retrieving pathway genes from KEGG pathway database A. K. M. Azad School of Biotechnology and Biomolecular Sciences, University of NSW, Chancellery Walk, Kensington, 2033, Australia Abstract Pathway analysis is a very important aspect in computational systems bi- ology as it serves as a crucial component in many computational pipelines. KEGG is one of the prominent databases that host pathway information associated with various organisms. In any pathway analysis pipelines, it is also important to collect and organize the pathway constituent genes for which a tool to automatically retrieve that would be a useful one to the practitioners. In this article, I present KPGminer, a tool that retrieves the constituent genes in KEGG pathways for various organisms and or- ganizes that information suitable for many downstream pathway analysis pipelines. We exploited several KEGG web services using REST APIs, par- ticularly GET and LIST methods to request for the information retrieval which is available for developers. Moreover, KPGminer can operate both for a particular pathway (single mode) or multiple pathways (batch mode). Next, we designed a crawler to extract necessary information from the re- sponse and generated outputs accordingly. KPGminer brings several key features including organism-specific and pathway-specific extraction of path- way genes from KEGG and always up-to-date information. -

Downregulated Developmental Processes in the Postnatal Right
www.nature.com/cddiscovery ARTICLE OPEN Downregulated developmental processes in the postnatal right ventricle under the influence of a volume overload ✉ ✉ ✉ Chunxia Zhou1,6, Sijuan Sun2,6, Mengyu Hu3, Yingying Xiao1, Xiafeng Yu1 , Lincai Ye 1,4,5 and Lisheng Qiu 1 © The Author(s) 2021 The molecular atlas of postnatal mouse ventricular development has been made available and cardiac regeneration is documented to be a downregulated process. The right ventricle (RV) differs from the left ventricle. How volume overload (VO), a common pathologic state in children with congenital heart disease, affects the downregulated processes of the RV is currently unclear. We created a fistula between the abdominal aorta and inferior vena cava on postnatal day 7 (P7) using a mouse model to induce a prepubertal RV VO. RNAseq analysis of RV (from postnatal day 14 to 21) demonstrated that angiogenesis was the most enriched gene ontology (GO) term in both the sham and VO groups. Regulation of the mitotic cell cycle was the second-most enriched GO term in the VO group but it was not in the list of enriched GO terms in the sham group. In addition, the number of Ki67-positive cardiomyocytes increased approximately 20-fold in the VO group compared to the sham group. The intensity of the vascular endothelial cells also changed dramatically over time in both groups. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the downregulated transcriptome revealed that the peroxisome proliferators-activated receptor (PPAR) signaling pathway was replaced by the cell cycle in the top-20 enriched KEGG terms because of the VO. -

Transcriptome Analysis of the Regulatory Mechanism of Foxo on Wing Dimorphism in the Brown Planthopper, Nilaparvata Lugens (Hemiptera: Delphacidae)
insects Article Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae) Nan Xu 1, Sheng-Fei Wei 1 and Hai-Jun Xu 1,2,3,* 1 State Key Laboratory of Rice Biology, Zhejiang University, Hangzhou 310058, China; [email protected] (N.X.); [email protected] (S.-F.W.) 2 Ministry of Agriculture Key Laboratory of Molecular Biology of Crop Pathogens and Insect Pests, Zhejiang University, Hangzhou 310058, China 3 Institute of Insect Sciences, Zhejiang University, Hangzhou 310058, China * Correspondence: [email protected]; Tel.: +86-571-88982996 Simple Summary: The brown planthopper (BPH) Nilaparvata lugens can develop into either long- winged or short-winged adults depending on environmental stimuli received during larval stages. The transcription factor NlFoxO serves as a key regulator determining alternative wing morphs in BPH, but the underlying molecular mechanism is largely unknown. Here, we investigated the transcriptomic profile of forewing and hindwing buds across the 5th-instar stage, the wing-morph decision stage. Our results indicated that NlFoxO modulated the developmental plasticity of wing buds mainly by regulating the expression of cell proliferation-associated genes. Abstract: The brown planthopper (BPH), Nilaparvata lugens, can develop into either short-winged (SW) or long-winged (LW) adults according to environmental conditions, and has long served as a model organism for exploring the mechanisms of wing polyphenism in insects. The transcription Citation: Xu, N.; Wei, S.-F.; Xu, H.-J. factor NlFoxO acts as a master regulator that directs the development of either SW or LW morphs, Transcriptome Analysis of the but the underlying molecular mechanism is largely unknown. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

"Using the KEGG Database Resource". In: Current Protocols in Bioinformatics
Using the KEGG Database Resource UNIT 1.12 KEGG (Kyoto Encyclopedia of Genes and Genomes) is a bioinformatics resource for understanding biological function from a genomic perspective. It is a multispecies, integrated resource consisting of genomic, chemical, and network information with cross-references to numerous outside databases and containing a complete set of building blocks (genes and molecules) and wiring diagrams (biological pathways) to represent cellular functions (Kanehisa et al., 2004). In this unit, protocols are described for using the five major KEGG resources: PATHWAY, GENES, SSDB, EXPRESSION, and LIGAND. The KEGG PATHWAY database (see Basic Protocols 1 to 4) consists of a user-friendly tool for analyzing the network of protein and small-molecule interactions that occur in the cells of various organisms. KEGG GENES (see Basic Protocols 5 and 6) provides access to the collection of gene data organized so as to be accessible via text searches, from the PATHWAY database, or via cross-species orthology searches. The KEGG Sequence Similiarity Database (SSDB; see Basic Protocols 7 to 9) consists of a precomputed database of all-versus-all Smith- Waterman similarity scores among all genes in KEGG GENES, enabling relationships between homologs to be easily visualized on the pathway and genome maps or viewed as clusters of orthologous genes. The KEGG EXPRESSION database (see Basic Protocols 10 to 14) contains both data and tools for analyzing gene expression data. User-defined data such as microarray experiments may also be uploaded for analysis using the tools available in KEGG EXPRESSION. Finally, KEGG LIGAND (see Basic Protocols 16 to 19) is a database of small molecules, their structures, and information relating to the enzymes that act on them. -

KEGG: Kyoto Encyclopedia of Genes and Genomes Hiroyuki Ogata, Susumu Goto, Kazushige Sato, Wataru Fujibuchi, Hidemasa Bono and Minoru Kanehisa*
1999 Oxford University Press Nucleic Acids Research, 1999, Vol. 27, No. 1 29–34 KEGG: Kyoto Encyclopedia of Genes and Genomes Hiroyuki Ogata, Susumu Goto, Kazushige Sato, Wataru Fujibuchi, Hidemasa Bono and Minoru Kanehisa* Institute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan Received September 8, 1998; Revised September 22, 1998; Accepted October 14, 1998 ABSTRACT The basic concepts of KEGG (1) and underlying informatics technologies (2,3) have already been published. KEGG is tightly Kyoto Encyclopedia of Genes and Genomes (KEGG) is integrated with the LIGAND chemical database for enzyme a knowledge base for systematic analysis of gene reactions (4,5) as well as with most of the major molecular functions in terms of the networks of genes and biology databases by the DBGET/LinkDB system (6) under the molecules. The major component of KEGG is the Japanese GenomeNet service (7). The database organization PATHWAY database that consists of graphical dia- efforts require extensive analyses of completely sequenced grams of biochemical pathways including most of the genomes, as exemplified by the analyses of metabolic pathways known metabolic pathways and some of the known (8) and ABC transport systems (9). In this article, we describe the regulatory pathways. The pathway information is also current status of the KEGG databases and discuss the use of represented by the ortholog group tables summarizing KEGG for functional genomics. orthologous and paralogous gene groups among different organisms. KEGG maintains the GENES OBJECTIVES OF KEGG database for the gene catalogs of all organisms with complete genomes and selected organisms with In May 1995, we initiated the KEGG project under the Human partial genomes, which are continuously re-annotated, Genome Program of the Ministry of Education, Science, Sports as well as the LIGAND database for chemical com- and Culture in Japan.