Automated Interpretation of Metabolic Capacity from Genome and Metagenome Sequences
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Method to Infer Changed Activity of Metabolic Function from Transcript Profiles
ModeScore: A Method to Infer Changed Activity of Metabolic Function from Transcript Profiles Andreas Hoppe and Hermann-Georg Holzhütter Charité University Medicine Berlin, Institute for Biochemistry, Computational Systems Biochemistry Group [email protected] Abstract Genome-wide transcript profiles are often the only available quantitative data for a particular perturbation of a cellular system and their interpretation with respect to the metabolism is a major challenge in systems biology, especially beyond on/off distinction of genes. We present a method that predicts activity changes of metabolic functions by scoring reference flux distributions based on relative transcript profiles, providing a ranked list of most regulated functions. Then, for each metabolic function, the involved genes are ranked upon how much they represent a specific regulation pattern. Compared with the naïve pathway-based approach, the reference modes can be chosen freely, and they represent full metabolic functions, thus, directly provide testable hypotheses for the metabolic study. In conclusion, the novel method provides promising functions for subsequent experimental elucidation together with outstanding associated genes, solely based on transcript profiles. 1998 ACM Subject Classification J.3 Life and Medical Sciences Keywords and phrases Metabolic network, expression profile, metabolic function Digital Object Identifier 10.4230/OASIcs.GCB.2012.1 1 Background The comprehensive study of the cell’s metabolism would include measuring metabolite concentrations, reaction fluxes, and enzyme activities on a large scale. Measuring fluxes is the most difficult part in this, for a recent assessment of techniques, see [31]. Although mass spectrometry allows to assess metabolite concentrations in a more comprehensive way, the larger the set of potential metabolites, the more difficult [8]. -

The Kyoto Encyclopedia of Genes and Genomes (KEGG)
Kyoto Encyclopedia of Genes and Genome Minoru Kanehisa Institute for Chemical Research, Kyoto University HFSPO Workshop, Strasbourg, November 18, 2016 The KEGG Databases Category Database Content PATHWAY KEGG pathway maps Systems information BRITE BRITE functional hierarchies MODULE KEGG modules KO (KEGG ORTHOLOGY) KO groups for functional orthologs Genomic information GENOME KEGG organisms, viruses and addendum GENES / SSDB Genes and proteins / sequence similarity COMPOUND Chemical compounds GLYCAN Glycans Chemical information REACTION / RCLASS Reactions / reaction classes ENZYME Enzyme nomenclature DISEASE Human diseases DRUG / DGROUP Drugs / drug groups Health information ENVIRON Health-related substances (KEGG MEDICUS) JAPIC Japanese drug labels DailyMed FDA drug labels 12 manually curated original DBs 3 DBs taken from outside sources and given original annotations (GENOME, GENES, ENZYME) 1 computationally generated DB (SSDB) 2 outside DBs (JAPIC, DailyMed) KEGG is widely used for functional interpretation and practical application of genome sequences and other high-throughput data KO PATHWAY GENOME BRITE DISEASE GENES MODULE DRUG Genome Molecular High-level Practical Metagenome functions functions applications Transcriptome etc. Metabolome Glycome etc. COMPOUND GLYCAN REACTION Funding Annual budget Period Funding source (USD) 1995-2010 Supported by 10+ grants from Ministry of Education, >2 M Japan Society for Promotion of Science (JSPS) and Japan Science and Technology Agency (JST) 2011-2013 Supported by National Bioscience Database Center 0.8 M (NBDC) of JST 2014-2016 Supported by NBDC 0.5 M 2017- ? 1995 KEGG website made freely available 1997 KEGG FTP site made freely available 2011 Plea to support KEGG KEGG FTP academic subscription introduced 1998 First commercial licensing Contingency Plan 1999 Pathway Solutions Inc. -

3 13437143.Pdf
Title Integrative Annotation of 21,037 Human Genes Validated by Full-Length cDNA Clones Imanishi, Tadashi; Itoh, Takeshi; Suzuki, Yutaka; O'Donovan, Claire; Fukuchi, Satoshi; Koyanagi, Kanako O.; Barrero, Roberto A.; Tamura, Takuro; Yamaguchi-Kabata, Yumi; Tanino, Motohiko; Yura, Kei; Miyazaki, Satoru; Ikeo, Kazuho; Homma, Keiichi; Kasprzyk, Arek; Nishikawa, Tetsuo; Hirakawa, Mika; Thierry-Mieg, Jean; Thierry-Mieg, Danielle; Ashurst, Jennifer; Jia, Libin; Nakao, Mitsuteru; Thomas, Michael A.; Mulder, Nicola; Karavidopoulou, Youla; Jin, Lihua; Kim, Sangsoo; Yasuda, Tomohiro; Lenhard, Boris; Eveno, Eric; Suzuki, Yoshiyuki; Yamasaki, Chisato; Takeda, Jun-ichi; Gough, Craig; Hilton, Phillip; Fujii, Yasuyuki; Sakai, Hiroaki; Tanaka, Susumu; Amid, Clara; Bellgard, Matthew; Bonaldo, Maria de Fatima; Bono, Hidemasa; Bromberg, Susan K.; Brookes, Anthony J.; Bruford, Elspeth; Carninci, Piero; Chelala, Claude; Couillault, Christine; Souza, Sandro J. de; Debily, Marie-Anne; Devignes, Marie-Dominique; Dubchak, Inna; Endo, Toshinori; Estreicher, Anne; Eyras, Eduardo; Fukami-Kobayashi, Kaoru; R. Gopinath, Gopal; Graudens, Esther; Hahn, Yoonsoo; Han, Michael; Han, Ze-Guang; Hanada, Kousuke; Hanaoka, Hideki; Harada, Erimi; Hashimoto, Katsuyuki; Hinz, Ursula; Hirai, Momoki; Hishiki, Teruyoshi; Hopkinson, Ian; Imbeaud, Sandrine; Inoko, Hidetoshi; Kanapin, Alexander; Kaneko, Yayoi; Kasukawa, Takeya; Kelso, Janet; Kersey, Author(s) Paul; Kikuno, Reiko; Kimura, Kouichi; Korn, Bernhard; Kuryshev, Vladimir; Makalowska, Izabela; Makino, Takashi; Mano, Shuhei; -

Precise Generation of Systems Biology Models from KEGG Pathways Clemens Wrzodek*,Finjabuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell
Wrzodek et al. BMC Systems Biology 2013, 7:15 http://www.biomedcentral.com/1752-0509/7/15 METHODOLOGY ARTICLE OpenAccess Precise generation of systems biology models from KEGG pathways Clemens Wrzodek*,FinjaBuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell Abstract Background: The KEGG PATHWAY database provides a plethora of pathways for a diversity of organisms. All pathway components are directly linked to other KEGG databases, such as KEGG COMPOUND or KEGG REACTION. Therefore, the pathways can be extended with an enormous amount of information and provide a foundation for initial structural modeling approaches. As a drawback, KGML-formatted KEGG pathways are primarily designed for visualization purposes and often omit important details for the sake of a clear arrangement of its entries. Thus, a direct conversion into systems biology models would produce incomplete and erroneous models. Results: Here, we present a precise method for processing and converting KEGG pathways into initial metabolic and signaling models encoded in the standardized community pathway formats SBML (Levels 2 and 3) and BioPAX (Levels 2 and 3). This method involves correcting invalid or incomplete KGML content, creating complete and valid stoichiometric reactions, translating relations to signaling models and augmenting the pathway content with various information, such as cross-references to Entrez Gene, OMIM, UniProt ChEBI, and many more. Finally, we compare several existing conversion tools for KEGG pathways and show that the conversion from KEGG to BioPAX does not involve a loss of information, whilst lossless translations to SBML can only be performed using SBML Level 3, including its recently proposed qualitative models and groups extension packages. -

Genbank by Walter B
GenBank by Walter B. Goad o understand the significance of the information stored in memory—DNA—to the stuff of activity—proteins. The idea that GenBank, you need to know a little about molecular somehow the bases in DNA determine the amino acids in proteins genetics. What that field deals with is self-replication—the had been around for some time. In fact, George Gamow suggested in T process unique to life—and mutation and 1954, after learning about the structure proposed for DNA, that a recombination—the processes responsible for evolution-at the triplet of bases corresponded to an amino acid. That suggestion was fundamental level of the genes in DNA. This approach of working shown to be true, and by 1965 most of the genetic code had been from the blueprint, so to speak, of a living system is very powerful, deciphered. Also worked out in the ’60s were many details of what and studies of many other aspects of life—the process of learning, for Crick called the central dogma of molecular genetics—the now example—are now utilizing molecular genetics. firmly established fact that DNA is not translated directly to proteins Molecular genetics began in the early ’40s and was at first but is first transcribed to messenger RNA. This molecule, a nucleic controversial because many of the people involved had been trained acid like DNA, then serves as the template for protein synthesis. in the physical sciences rather than the biological sciences, and yet These great advances prompted a very distinguished molecular they were answering questions that biologists had been asking for geneticist to predict, in 1969, that biology was just about to end since years. -

Integrative Annotation of 21,037 Human Genes Validated by Full-Length Cdna Clones
Title Integrative Annotation of 21,037 Human Genes Validated by Full-Length cDNA Clones Imanishi, Tadashi; Itoh, Takeshi; Suzuki, Yutaka; O'Donovan, Claire; Fukuchi, Satoshi; Koyanagi, Kanako O.; Barrero, Roberto A.; Tamura, Takuro; Yamaguchi-Kabata, Yumi; Tanino, Motohiko; Yura, Kei; Miyazaki, Satoru; Ikeo, Kazuho; Homma, Keiichi; Kasprzyk, Arek; Nishikawa, Tetsuo; Hirakawa, Mika; Thierry-Mieg, Jean; Thierry-Mieg, Danielle; Ashurst, Jennifer; Jia, Libin; Nakao, Mitsuteru; Thomas, Michael A.; Mulder, Nicola; Karavidopoulou, Youla; Jin, Lihua; Kim, Sangsoo; Yasuda, Tomohiro; Lenhard, Boris; Eveno, Eric; Suzuki, Yoshiyuki; Yamasaki, Chisato; Takeda, Jun-ichi; Gough, Craig; Hilton, Phillip; Fujii, Yasuyuki; Sakai, Hiroaki; Tanaka, Susumu; Amid, Clara; Bellgard, Matthew; Bonaldo, Maria de Fatima; Bono, Hidemasa; Bromberg, Susan K.; Brookes, Anthony J.; Bruford, Elspeth; Carninci, Piero; Chelala, Claude; Couillault, Christine; Souza, Sandro J. de; Debily, Marie-Anne; Devignes, Marie-Dominique; Dubchak, Inna; Endo, Toshinori; Estreicher, Anne; Eyras, Eduardo; Fukami-Kobayashi, Kaoru; R. Gopinath, Gopal; Graudens, Esther; Hahn, Yoonsoo; Han, Michael; Han, Ze-Guang; Hanada, Kousuke; Hanaoka, Hideki; Harada, Erimi; Hashimoto, Katsuyuki; Hinz, Ursula; Hirai, Momoki; Hishiki, Teruyoshi; Hopkinson, Ian; Imbeaud, Sandrine; Inoko, Hidetoshi; Kanapin, Alexander; Kaneko, Yayoi; Kasukawa, Takeya; Kelso, Janet; Kersey, Author(s) Paul; Kikuno, Reiko; Kimura, Kouichi; Korn, Bernhard; Kuryshev, Vladimir; Makalowska, Izabela; Makino, Takashi; Mano, Shuhei; -
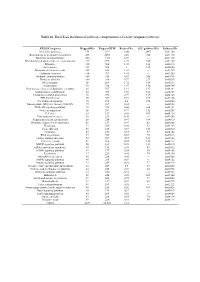
Table 1S. the KEGG Biochemical Pathways Categorization of LA Lily Unigenes Pathways
Table 1S. The KEGG biochemical pathways categorization of LA lily unigenes pathways. KEGG Categories Mapped-KO Unigene-NUM Ratio of No. ALL pathway KO Pathway-ID Metabolic pathways 954 4337 8.66 2067 ko01100 Biosynthesis of secondary metabolites 403 2285 4.56 720 ko01110 Biosynthesis of antibiotics 206 1147 2.29 --- ko01130 Microbial metabolism in diverse environments 157 995 1.99 720 ko01120 Ribosome 122 504 1.01 142 ko03010 Spliceosome 107 502 1 115 ko03040 Biosynthesis of amino acids 105 614 1.23 --- ko01230 Carbon metabolism 104 707 1.41 --- ko01200 Oxidative phosphorylation 100 335 0.67 206 ko00190 Purine metabolism 100 386 0.77 237 ko00230 RNA transport 98 861 1.72 134 ko03013 Endocytosis 92 736 1.47 138 ko04144 Protein processing in endoplasmic reticulum 88 567 1.13 137 ko04141 homologous recombination 84 933 1.86 144 ko05169 Ubiquitin mediated proteolysis 78 396 0.79 119 ko04120 HTLV-I infection 76 367 0.73 199 ko05166 Pyrimidine metabolism 76 298 0.6 150 ko00240 Non-alcoholic fatty liver disease (NAFLD) 72 209 0.42 --- ko04932 PI3K-Akt signaling pathway 71 328 0.66 226 ko04151 Viral carcinogenesis 68 387 0.77 132 ko05203 Cell cycle 63 339 0.68 103 ko04110 Proteoglycans in cancer 58 229 0.46 --- ko05205 Regulation of actin cytoskeleton 58 234 0.47 144 ko04810 Ribosome biogenesis in eukaryotes 56 237 0.47 82 ko03008 Phagosome 54 280 0.56 93 ko04145 Focal adhesion 53 185 0.37 133 ko04510 Lysosome 53 253 0.51 99 ko04142 RNA degradation 53 305 0.61 70 ko03018 Herpes simplex infection 52 257 0.51 121 ko05168 Cell cycle - yeast 51 260 -

I S C B N E W S L E T T
ISCB NEWSLETTER FOCUS ISSUE {contents} President’s Letter 2 Member Involvement Encouraged Register for ISMB 2002 3 Registration and Tutorial Update Host ISMB 2004 or 2005 3 David Baker 4 2002 Overton Prize Recipient Overton Endowment 4 ISMB 2002 Committees 4 ISMB 2002 Opportunities 5 Sponsor and Exhibitor Benefits Best Paper Award by SGI 5 ISMB 2002 SIGs 6 New Program for 2002 ISMB Goes Down Under 7 Planning Underway for 2003 Hot Jobs! Top Companies! 8 ISMB 2002 Job Fair ISCB Board Nominations 8 Bioinformatics Pioneers 9 ISMB 2002 Keynote Speakers Invited Editorial 10 Anna Tramontano: Bioinformatics in Europe Software Recommendations11 ISCB Software Statement volume 5. issue 2. summer 2002 Community Development 12 ISCB’s Regional Affiliates Program ISCB Staff Introduction 12 Fellowship Recipients 13 Awardees at RECOMB 2002 Events and Opportunities 14 Bioinformatics events world wide INTERNATIONAL SOCIETY FOR COMPUTATIONAL BIOLOGY A NOTE FROM ISCB PRESIDENT This newsletter is packed with information on development and dissemination of bioinfor- the ISMB2002 conference. With over 200 matics. Issues arise from recommendations paper submissions and over 500 poster submis- made by the Society’s committees, Board of sions, the conference promises to be a scientific Directors, and membership at large. Important feast. On behalf of the ISCB’s Directors, staff, issues are defined as motions and are discussed EXECUTIVE COMMITTEE and membership, I would like to thank the by the Board of Directors on a bi-monthly Philip E. Bourne, Ph.D., President organizing committee, local organizing com- teleconference. Motions that pass are enacted Michael Gribskov, Ph.D., mittee, and program committee for their hard by the Executive Committee which also serves Vice President work preparing for the conference. -

UCLA UCLA Electronic Theses and Dissertations
UCLA UCLA Electronic Theses and Dissertations Title Bipartite Network Community Detection: Development and Survey of Algorithmic and Stochastic Block Model Based Methods Permalink https://escholarship.org/uc/item/0tr9j01r Author Sun, Yidan Publication Date 2021 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA Los Angeles Bipartite Network Community Detection: Development and Survey of Algorithmic and Stochastic Block Model Based Methods A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Statistics by Yidan Sun 2021 © Copyright by Yidan Sun 2021 ABSTRACT OF THE DISSERTATION Bipartite Network Community Detection: Development and Survey of Algorithmic and Stochastic Block Model Based Methods by Yidan Sun Doctor of Philosophy in Statistics University of California, Los Angeles, 2021 Professor Jingyi Li, Chair In a bipartite network, nodes are divided into two types, and edges are only allowed to connect nodes of different types. Bipartite network clustering problems aim to identify node groups with more edges between themselves and fewer edges to the rest of the network. The approaches for community detection in the bipartite network can roughly be classified into algorithmic and model-based methods. The algorithmic methods solve the problem either by greedy searches in a heuristic way or optimizing based on some criteria over all possible partitions. The model-based methods fit a generative model to the observed data and study the model in a statistically principled way. In this dissertation, we mainly focus on bipartite clustering under two scenarios: incorporation of node covariates and detection of mixed membership communities. -

A Tool for Retrieving Pathway Genes from KEGG Pathway Database
bioRxiv preprint doi: https://doi.org/10.1101/416131; this version posted September 13, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. KPGminer: A tool for retrieving pathway genes from KEGG pathway database A. K. M. Azad School of Biotechnology and Biomolecular Sciences, University of NSW, Chancellery Walk, Kensington, 2033, Australia Abstract Pathway analysis is a very important aspect in computational systems bi- ology as it serves as a crucial component in many computational pipelines. KEGG is one of the prominent databases that host pathway information associated with various organisms. In any pathway analysis pipelines, it is also important to collect and organize the pathway constituent genes for which a tool to automatically retrieve that would be a useful one to the practitioners. In this article, I present KPGminer, a tool that retrieves the constituent genes in KEGG pathways for various organisms and or- ganizes that information suitable for many downstream pathway analysis pipelines. We exploited several KEGG web services using REST APIs, par- ticularly GET and LIST methods to request for the information retrieval which is available for developers. Moreover, KPGminer can operate both for a particular pathway (single mode) or multiple pathways (batch mode). Next, we designed a crawler to extract necessary information from the re- sponse and generated outputs accordingly. KPGminer brings several key features including organism-specific and pathway-specific extraction of path- way genes from KEGG and always up-to-date information. -

Downregulated Developmental Processes in the Postnatal Right
www.nature.com/cddiscovery ARTICLE OPEN Downregulated developmental processes in the postnatal right ventricle under the influence of a volume overload ✉ ✉ ✉ Chunxia Zhou1,6, Sijuan Sun2,6, Mengyu Hu3, Yingying Xiao1, Xiafeng Yu1 , Lincai Ye 1,4,5 and Lisheng Qiu 1 © The Author(s) 2021 The molecular atlas of postnatal mouse ventricular development has been made available and cardiac regeneration is documented to be a downregulated process. The right ventricle (RV) differs from the left ventricle. How volume overload (VO), a common pathologic state in children with congenital heart disease, affects the downregulated processes of the RV is currently unclear. We created a fistula between the abdominal aorta and inferior vena cava on postnatal day 7 (P7) using a mouse model to induce a prepubertal RV VO. RNAseq analysis of RV (from postnatal day 14 to 21) demonstrated that angiogenesis was the most enriched gene ontology (GO) term in both the sham and VO groups. Regulation of the mitotic cell cycle was the second-most enriched GO term in the VO group but it was not in the list of enriched GO terms in the sham group. In addition, the number of Ki67-positive cardiomyocytes increased approximately 20-fold in the VO group compared to the sham group. The intensity of the vascular endothelial cells also changed dramatically over time in both groups. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the downregulated transcriptome revealed that the peroxisome proliferators-activated receptor (PPAR) signaling pathway was replaced by the cell cycle in the top-20 enriched KEGG terms because of the VO. -

Transcriptome Analysis of the Regulatory Mechanism of Foxo on Wing Dimorphism in the Brown Planthopper, Nilaparvata Lugens (Hemiptera: Delphacidae)
insects Article Transcriptome Analysis of the Regulatory Mechanism of FoxO on Wing Dimorphism in the Brown Planthopper, Nilaparvata lugens (Hemiptera: Delphacidae) Nan Xu 1, Sheng-Fei Wei 1 and Hai-Jun Xu 1,2,3,* 1 State Key Laboratory of Rice Biology, Zhejiang University, Hangzhou 310058, China; [email protected] (N.X.); [email protected] (S.-F.W.) 2 Ministry of Agriculture Key Laboratory of Molecular Biology of Crop Pathogens and Insect Pests, Zhejiang University, Hangzhou 310058, China 3 Institute of Insect Sciences, Zhejiang University, Hangzhou 310058, China * Correspondence: [email protected]; Tel.: +86-571-88982996 Simple Summary: The brown planthopper (BPH) Nilaparvata lugens can develop into either long- winged or short-winged adults depending on environmental stimuli received during larval stages. The transcription factor NlFoxO serves as a key regulator determining alternative wing morphs in BPH, but the underlying molecular mechanism is largely unknown. Here, we investigated the transcriptomic profile of forewing and hindwing buds across the 5th-instar stage, the wing-morph decision stage. Our results indicated that NlFoxO modulated the developmental plasticity of wing buds mainly by regulating the expression of cell proliferation-associated genes. Abstract: The brown planthopper (BPH), Nilaparvata lugens, can develop into either short-winged (SW) or long-winged (LW) adults according to environmental conditions, and has long served as a model organism for exploring the mechanisms of wing polyphenism in insects. The transcription Citation: Xu, N.; Wei, S.-F.; Xu, H.-J. factor NlFoxO acts as a master regulator that directs the development of either SW or LW morphs, Transcriptome Analysis of the but the underlying molecular mechanism is largely unknown.