Pathway Analysis in Metabolomics: Pitfalls and Best Practice for the Use of Over-Representation Analysis
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Kyoto Encyclopedia of Genes and Genomes (KEGG)
Kyoto Encyclopedia of Genes and Genome Minoru Kanehisa Institute for Chemical Research, Kyoto University HFSPO Workshop, Strasbourg, November 18, 2016 The KEGG Databases Category Database Content PATHWAY KEGG pathway maps Systems information BRITE BRITE functional hierarchies MODULE KEGG modules KO (KEGG ORTHOLOGY) KO groups for functional orthologs Genomic information GENOME KEGG organisms, viruses and addendum GENES / SSDB Genes and proteins / sequence similarity COMPOUND Chemical compounds GLYCAN Glycans Chemical information REACTION / RCLASS Reactions / reaction classes ENZYME Enzyme nomenclature DISEASE Human diseases DRUG / DGROUP Drugs / drug groups Health information ENVIRON Health-related substances (KEGG MEDICUS) JAPIC Japanese drug labels DailyMed FDA drug labels 12 manually curated original DBs 3 DBs taken from outside sources and given original annotations (GENOME, GENES, ENZYME) 1 computationally generated DB (SSDB) 2 outside DBs (JAPIC, DailyMed) KEGG is widely used for functional interpretation and practical application of genome sequences and other high-throughput data KO PATHWAY GENOME BRITE DISEASE GENES MODULE DRUG Genome Molecular High-level Practical Metagenome functions functions applications Transcriptome etc. Metabolome Glycome etc. COMPOUND GLYCAN REACTION Funding Annual budget Period Funding source (USD) 1995-2010 Supported by 10+ grants from Ministry of Education, >2 M Japan Society for Promotion of Science (JSPS) and Japan Science and Technology Agency (JST) 2011-2013 Supported by National Bioscience Database Center 0.8 M (NBDC) of JST 2014-2016 Supported by NBDC 0.5 M 2017- ? 1995 KEGG website made freely available 1997 KEGG FTP site made freely available 2011 Plea to support KEGG KEGG FTP academic subscription introduced 1998 First commercial licensing Contingency Plan 1999 Pathway Solutions Inc. -

Precise Generation of Systems Biology Models from KEGG Pathways Clemens Wrzodek*,Finjabuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell
Wrzodek et al. BMC Systems Biology 2013, 7:15 http://www.biomedcentral.com/1752-0509/7/15 METHODOLOGY ARTICLE OpenAccess Precise generation of systems biology models from KEGG pathways Clemens Wrzodek*,FinjaBuchel,¨ Manuel Ruff, Andreas Drager¨ and Andreas Zell Abstract Background: The KEGG PATHWAY database provides a plethora of pathways for a diversity of organisms. All pathway components are directly linked to other KEGG databases, such as KEGG COMPOUND or KEGG REACTION. Therefore, the pathways can be extended with an enormous amount of information and provide a foundation for initial structural modeling approaches. As a drawback, KGML-formatted KEGG pathways are primarily designed for visualization purposes and often omit important details for the sake of a clear arrangement of its entries. Thus, a direct conversion into systems biology models would produce incomplete and erroneous models. Results: Here, we present a precise method for processing and converting KEGG pathways into initial metabolic and signaling models encoded in the standardized community pathway formats SBML (Levels 2 and 3) and BioPAX (Levels 2 and 3). This method involves correcting invalid or incomplete KGML content, creating complete and valid stoichiometric reactions, translating relations to signaling models and augmenting the pathway content with various information, such as cross-references to Entrez Gene, OMIM, UniProt ChEBI, and many more. Finally, we compare several existing conversion tools for KEGG pathways and show that the conversion from KEGG to BioPAX does not involve a loss of information, whilst lossless translations to SBML can only be performed using SBML Level 3, including its recently proposed qualitative models and groups extension packages. -

S41598-018-25035-1.Pdf
www.nature.com/scientificreports OPEN An Innovative Approach for The Integration of Proteomics and Metabolomics Data In Severe Received: 23 October 2017 Accepted: 9 April 2018 Septic Shock Patients Stratifed for Published: xx xx xxxx Mortality Alice Cambiaghi1, Ramón Díaz2, Julia Bauzá Martinez2, Antonia Odena2, Laura Brunelli3, Pietro Caironi4,5, Serge Masson3, Giuseppe Baselli1, Giuseppe Ristagno 3, Luciano Gattinoni6, Eliandre de Oliveira2, Roberta Pastorelli3 & Manuela Ferrario 1 In this work, we examined plasma metabolome, proteome and clinical features in patients with severe septic shock enrolled in the multicenter ALBIOS study. The objective was to identify changes in the levels of metabolites involved in septic shock progression and to integrate this information with the variation occurring in proteins and clinical data. Mass spectrometry-based targeted metabolomics and untargeted proteomics allowed us to quantify absolute metabolites concentration and relative proteins abundance. We computed the ratio D7/D1 to take into account their variation from day 1 (D1) to day 7 (D7) after shock diagnosis. Patients were divided into two groups according to 28-day mortality. Three diferent elastic net logistic regression models were built: one on metabolites only, one on metabolites and proteins and one to integrate metabolomics and proteomics data with clinical parameters. Linear discriminant analysis and Partial least squares Discriminant Analysis were also implemented. All the obtained models correctly classifed the observations in the testing set. By looking at the variable importance (VIP) and the selected features, the integration of metabolomics with proteomics data showed the importance of circulating lipids and coagulation cascade in septic shock progression, thus capturing a further layer of biological information complementary to metabolomics information. -

A Metabolomics Approach to Pharmacotherapy Personalization
Journal of Personalized Medicine Review A Metabolomics Approach to Pharmacotherapy Personalization Elena E. Balashova *, Dmitry L. Maslov and Petr G. Lokhov Institute of Biomedical Chemistry, Pogodinskaya St. 10, Moscow 119121, Russia; [email protected] (D.L.M.); [email protected] (P.G.L.) * Correspondence: [email protected] Received: 29 June 2018; Accepted: 3 September 2018; Published: 5 September 2018 Abstract: The optimization of drug therapy according to the personal characteristics of patients is a perspective direction in modern medicine. One of the possible ways to achieve such personalization is through the application of “omics” technologies, including current, promising metabolomics methods. This review demonstrates that the analysis of pre-dose metabolite biofluid profiles allows clinicians to predict the effectiveness of a selected drug treatment for a given individual. In the review, it is also shown that the monitoring of post-dose metabolite profiles could allow clinicians to evaluate drug efficiency, the reaction of the host to the treatment, and the outcome of the therapy. A comparative description of pharmacotherapy personalization (pharmacogenomics, pharmacoproteomics, and therapeutic drug monitoring) and personalization based on the analysis of metabolite profiles for biofluids (pharmacometabolomics) is also provided. Keywords: pharmacometabolomics; metabolomics; pharmacogenomics; therapeutic drug monitoring; personalized medicine; mass spectrometry 1. Introduction The uniformity of the drug response or low inter-individual differences in drug response are commonly accepted tenets in the field of medicine. Almost all drugs are prescribed on the basis of this statement. This approach can be described as treatment of the “average patient” by “the average pill” or “one size fits all”. However, clinicians have long observed that the actual effectiveness of the pharmacotherapy may be variable. -

The Metabolomic Paradigm of Pharmacogenomics in Complex
ics: O om pe ol n b A a c t c e e M s s Cacabelos, Metabolomics 2012, 2:5 Metabolomics: Open Access DOI: 10.4172/2153-0769.1000e119 ISSN: 2153-0769 Editorial Open Access The Metabolomic Paradigm of Pharmacogenomics in Complex Disorders Ramón Cacabelos1,2* 1EuroEspes Biomedical Research Center, Institute for CNS Disorders and Genomic Medicine, EuroEspes Chair of Biotechnology and Genomics, 15165-Bergondo, Corunna, Spain 2President, World Association of Genomic Medicine, Spain Metabolomics represents the networking organization of multiple pathogenic genes usually converge in metabolomic networks leading biochemical pathways leading to a physiological function in living to specific pathogenic cascades responsible for disease phenotypes. organisms. The frontier between health and disease is likely to be the The genomics of the mechanism of action of drugs has so far been result of a fine-tuning equilibrium or disequilibrium, respectively, neglected by the scientific community, and consequently less than 5% between the genomic-transcriptomic-proteomic-metabolomic cascade of FDA-approved drugs, with a pharmacologically-defined mechanism and environmental factors and/or epigenetic phenomena. In recent of action, have been studied in order to evaluate whether mutations times, diverse metabolomic studies have emerged in medical science in the genes encoding receptors or enzymes may affect efficacy and to explain physiological and pathogenic events in several disciplines, safety issues. Pleiotropic genes, involved in multiple pathogenic events, -

Model-Based Integration of Genomics and Metabolomics Reveals SNP Functionality in Mycobacterium Tuberculosis
Model-based integration of genomics and metabolomics reveals SNP functionality in Mycobacterium tuberculosis Ove Øyåsa,b,1, Sonia Borrellc,d,1, Andrej Traunerc,d,1, Michael Zimmermanne, Julia Feldmannc,d, Thomas Liphardta,b, Sebastien Gagneuxc,d, Jörg Stellinga,b, Uwe Sauere, and Mattia Zampierie,2 aDepartment of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; bSIB Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland; cDepartment of Medical Parasitoloy and Infection Biology, Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; dUniversity of Basel, 4058 Basel, Switzerland; and eInstitute of Molecular Systems Biology, ETH Zurich, 8093 Zurich, Switzerland Edited by Ralph R. Isberg, Tufts University School of Medicine, Boston, MA, and approved March 2, 2020 (received for review September 12, 2019) Human tuberculosis is caused by members of the Mycobacterium infection of macrophages (29–32). Beyond analyses of individual tuberculosis complex (MTBC) that vary in virulence and transmis- laboratory strains, however, no systematic characterization and sibility. While genome-wide association studies have uncovered comparative analysis of intrinsic metabolic differences across several mutations conferring drug resistance, much less is known human-adapted MTBC clinical strains has been performed. about the factors underlying other bacterial phenotypes. Variation If the metabolic and other phenotypic diversity between in the outcome of tuberculosis infection and diseases has been MTBC strains contributes to and modulates pathogenicity, an attributed primarily to patient and environmental factors, but obvious question is: Which elements of the limited genetic di- recent evidence indicates an additional role for the genetic diver- versity in the MTBC are responsible for phenotypic strain di- sity among MTBC clinical strains. -

M. Tomita T. Nishioka (Eds.) Metabolomics the Frontier of Systems Biology
M. Tomita T. Nishioka (Eds.) Metabolomics The Frontier of Systems Biology M. Tomita, T. Nishioka (Eds.) Metabolomics The Frontier of Systems Biology With 112 Figures, Including 4 in Color Springer Masaru Tomita, Ph.D. Professor and Director Institute for Advanced Biosciences Keio University Tsuruoka 997-0035, Japan Takaaki Nishioka, Ph.D. Professor Graduate School of Agricuhure Kyoto University Kyoto 606-8502, Japan This book is based on the Japanese original, M. Tomita, T. Nishioka (Eds.), Metabolome Kenkyu no Saizensen, Springer-Verlag Tokyo, 2003. Library of Congress Control Number: 2005928331 ISBN 4-431-25121-9 Springer-Verlag Tokyo Berlin Heidelberg New York This work is subject to copyright. All rights are reserved, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in other ways, and storage in data banks. The use of registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. Product liability: The publishers cannot guarantee the accuracy of any information about dosage and application contained in this book. In every individual case the user must check such information by consulting the relevant literature. Springer is a part of Springer Science+Business Media springeronline.com © Springer-Vertag Tokyo 2005 Printed in Japan Typesetting: Camera-ready by the editor. Printing and binding: Nikkei Printing, Japan Printed on acid-free paper Preface The aim of this book is to review metabolomics research. -

Lipidomics and Metabolomics Service Gain Deeper Insights Into Exosomes
EXOSOMES LIPIDOMICS AND METABOLOMICS SERVICE GAIN DEEPER INSIGHTS INTO EXOSOMES SYSTEMBIO.COM/LIPIDOMICS HIGHLIGHTS What can lipidomics of exosomes tell you? n Discover novel circulating biomarkers Lipids are an important part of cellular physiology, and are increasingly being recognized for their importance in exosome biology as well. Exosomes were recently shown to have the highest lipid- n Learn more about exosome biology to-protein ratio of all classes of extracellular vesicles (1), with lipid content that both differs from n Send us your sample and receive data the parent cell the vesicles are shed from (2) and also changes as exosomes undergo a variety of in 4 - 6 weeks physiological processes (3). These unique lipid profiles can serve as novel circulating biomarkers, and recent evidence suggests that specific lipid species carried by exosomes can also modulate Service Overview the function of recipient cells (4). Whether you’re interested in With so much information revealed by lipid content, lipidomics studies of exosomes are a great way to identify lipid-based biomarkers and for understanding vesicle biogenesis and function (5). circulating biomarker discovery, basic exosome research, or other TUMOR MICROENVIRONMENT exosome-related studies, SBI’s CANCER CELLS CAFs EXOSOMES Exosome Lipidomics & Metabolomics Service helps you quickly and efficiently get the most information from your exosomes. Simply send Exosomes affect metabolism of cancer us your sample or purified exosomes cells A recent study by Zhao, et al, (6) and we’ll send back a report with demonstrated that exosomes from putative identifications, mass/charge patient-derived cancer-associated fibroblasts (CAFs) can reprogram EXOSOME ratios, and differential analysis (if UPTAKE the cellular machinery in cancer requested). -

IPA Analysis of Metabolomics Data Including Cross-Platform Integration with Transcriptomics Data from a Diabetic Mouse Model
IPA Analysis of Metabolomics Data Including Cross-Platform Integration with Transcriptomics Data from a Diabetic Mouse Model CONTENTS Overview ........................................................................................................................................................................ 3 Introduction ................................................................................................................................................................... 3 Metabolomics Background ........................................................................................................................................ 3 Integration of Metabolomic and Transcriptomic data .............................................................................................. 5 The Ingenuity Platform .............................................................................................................................................. 5 Case Study Background ............................................................................................................................................. 5 Methods ........................................................................................................................................................................ 6 Establishing a Metabolite profile for db/db mice and identification of exploratory biomarkers .............................. 6 IPA-Metabolomics™ Analysis: ................................................................................................................................... -

Proteomics and Metabolomics: the Final Frontier of Nutrition Research 71
SIGHT AND LIFE | VOL. 29(1) | 2015 PROTEOMICS AND METABOLOMICS: THE FINAL FRONTIER OF NUTRITION RESEARCH 71 Proteomics and Metabolomics: The Final Frontier of Nutrition Research Richard D Semba RNA editing, RNA splicing, post-translational modifications, and Wilmer Eye Institute, Johns Hopkins University School protein degradation; the proteome does not strictly reflect the of Medicine, Baltimore, Maryland, USA genome. Proteins function as enzymes, hormones, receptors, immune mediators, structure, transporters, and modulators of cell communication and signaling. The metabolome consists Introduction of amino acids, amines, peptides, sugars, oligonucleotides, ke- Revolutionary new technologies allow us to penetrate scientific tones, aldehydes, lipids, steroids, vitamins, and other molecules. frontiers and open vast new territories for discovery. In astrono- These metabolites reflect intrinsic chemical processes in cells my, the Hubble Space Telescope has facilitated an unprecedent- as well as environmental exposures such as diet and gut micro- ed view outwards, beyond our galaxy. Wherever the telescope is bial flora. The current Human Metabolome Database contains directed, scientists are making exciting new observations of the more than 40,000 entries7 – a number that is expected to grow deep universe. Another revolution is taking place in two fields quickly in the future. of “omics” research: proteomics and metabolomics. In contrast, The goals of proteomics include the detection of the diver- this view is directed inwards, towards the complexity of biologi- sity of proteins, their quantity, their isoforms, and the localiza- cal processes in living organisms. Proteomics is the study of the tion and interactions of proteins. The goals of metabolomics structure and function of proteins expressed by an organism. -
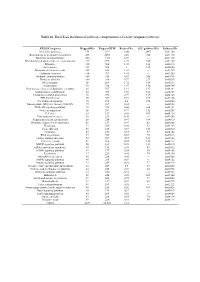
Table 1S. the KEGG Biochemical Pathways Categorization of LA Lily Unigenes Pathways
Table 1S. The KEGG biochemical pathways categorization of LA lily unigenes pathways. KEGG Categories Mapped-KO Unigene-NUM Ratio of No. ALL pathway KO Pathway-ID Metabolic pathways 954 4337 8.66 2067 ko01100 Biosynthesis of secondary metabolites 403 2285 4.56 720 ko01110 Biosynthesis of antibiotics 206 1147 2.29 --- ko01130 Microbial metabolism in diverse environments 157 995 1.99 720 ko01120 Ribosome 122 504 1.01 142 ko03010 Spliceosome 107 502 1 115 ko03040 Biosynthesis of amino acids 105 614 1.23 --- ko01230 Carbon metabolism 104 707 1.41 --- ko01200 Oxidative phosphorylation 100 335 0.67 206 ko00190 Purine metabolism 100 386 0.77 237 ko00230 RNA transport 98 861 1.72 134 ko03013 Endocytosis 92 736 1.47 138 ko04144 Protein processing in endoplasmic reticulum 88 567 1.13 137 ko04141 homologous recombination 84 933 1.86 144 ko05169 Ubiquitin mediated proteolysis 78 396 0.79 119 ko04120 HTLV-I infection 76 367 0.73 199 ko05166 Pyrimidine metabolism 76 298 0.6 150 ko00240 Non-alcoholic fatty liver disease (NAFLD) 72 209 0.42 --- ko04932 PI3K-Akt signaling pathway 71 328 0.66 226 ko04151 Viral carcinogenesis 68 387 0.77 132 ko05203 Cell cycle 63 339 0.68 103 ko04110 Proteoglycans in cancer 58 229 0.46 --- ko05205 Regulation of actin cytoskeleton 58 234 0.47 144 ko04810 Ribosome biogenesis in eukaryotes 56 237 0.47 82 ko03008 Phagosome 54 280 0.56 93 ko04145 Focal adhesion 53 185 0.37 133 ko04510 Lysosome 53 253 0.51 99 ko04142 RNA degradation 53 305 0.61 70 ko03018 Herpes simplex infection 52 257 0.51 121 ko05168 Cell cycle - yeast 51 260 -

A Tool for Retrieving Pathway Genes from KEGG Pathway Database
bioRxiv preprint doi: https://doi.org/10.1101/416131; this version posted September 13, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. KPGminer: A tool for retrieving pathway genes from KEGG pathway database A. K. M. Azad School of Biotechnology and Biomolecular Sciences, University of NSW, Chancellery Walk, Kensington, 2033, Australia Abstract Pathway analysis is a very important aspect in computational systems bi- ology as it serves as a crucial component in many computational pipelines. KEGG is one of the prominent databases that host pathway information associated with various organisms. In any pathway analysis pipelines, it is also important to collect and organize the pathway constituent genes for which a tool to automatically retrieve that would be a useful one to the practitioners. In this article, I present KPGminer, a tool that retrieves the constituent genes in KEGG pathways for various organisms and or- ganizes that information suitable for many downstream pathway analysis pipelines. We exploited several KEGG web services using REST APIs, par- ticularly GET and LIST methods to request for the information retrieval which is available for developers. Moreover, KPGminer can operate both for a particular pathway (single mode) or multiple pathways (batch mode). Next, we designed a crawler to extract necessary information from the re- sponse and generated outputs accordingly. KPGminer brings several key features including organism-specific and pathway-specific extraction of path- way genes from KEGG and always up-to-date information.