1,45%,562 UNITED SATES P All.‘ Bl If" Til?
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Safety Practices in Chemistry Laboratories
4/24/2012 Safety Practices in Chemistry Laboratory Sam Tung, HSE Specialist, CIH, RSO Health Safety and Environment Office, Hong Kong University of Science and Technology Contents 1. Introduction to Legislations related to Chemistry Laboratories 2. Introduction to Chemical Hazards 3. Criteria of Design for Laboratory Ventilation 4. Operations and Maintenance of Laboratory Fume hood 5. Case Studies of Laboratory Accidents 6. Handling of Chemical Spills 7. Emergency Response Equipment 1 4/24/2012 Importance of Health and Safety To protect your health and safety (and environment) To protect your colleagues’ and students’ health and safety Case Study: Laboratory Fire Kills UCLA Researcher A 23-year-old research assistant working at UCLA who was seriously burned in a lab fire in December 2008 recently died from her injuries. She was trying to transfer up to 2 ounces (~50ml) of t-butyl lithium (pyrophoric chemical), which was dissolved in pentane from one sealed container to another by a 50 ml syringe. The barrel of the syringe was either ejected or pulled out of the syringe, causing liquid to be released. A flash fire set her clothing ablaze and spread second- and third-degree burns over 43% of her body. 2 4/24/2012 Root Causes of the Accident Poor technique and improper method Use a 50ml syringe to transfer~50 ml pyrophoric chemical Should use a 100ml syringe Should use Cannula Method for transfer of pyrophoric chemical > 50 ml Lack of proper training No safety training record Lack of supervision No follow up actions had been -

Hazardous Material Inventory Statement
City of Brooklyn Park FIRE DEPARTMENT 5200 - 85th Avenue North Brooklyn Park MN 55443 Phone: (763)493-8020 Fax: (763) 493-8391 Hazardous Materials Inventory Statement Users Guide A separate inventory statement shall be provided for each building. An amended inventory statement shall be provided within 30 days of the storage of any hazardous materials or plastics that changes or adds a hazard class or which is sufficient in quantity to cause an increase in the quantity which exceeds 5 percent for any hazard class. The hazardous materials inventory statement shall list by hazard class categories. Each grouping shall provide the following information for each hazardous material listed for that group including a total quantity for each group of hazard class. 1. Hazard class. (See attached Hazardous Materials Categories Listing) 2. Common or trade name. 3. Chemical Abstract Service Number (CAS number) found in 29 Code of Federal Regulations (C.F.R.). 4. Whether the material is pure or a mixture, and whether the material is a solid, liquid or gas 5. Maximum aggregate quantity stored at any one time. 6. Maximum aggregate quantity In-Use (Open to atmosphere) at any one time. 7. Maximum aggregate quantity In-Use (Closed to atmosphere) at any one time. 8. Storage conditions related to the storage type, high-pile, encapsulated, non-encapsulated. Attached is a listing of categories that all materials need to be organized to. Definitions of these categories are also attached for your use. At the end of this packet are blank forms for completing this project. For questions regarding Hazardous Materials Inventory Statement contact the Fire Department at 763-493-8020. -

Synthesis and Properties of Simple and Complex Salts Of
PhD Theses SYNTHESIS AND PROPERTIES OF SIMPLE AND COMPLEX SALTS OF PERMANGANIC ACID Kótai László Institute of Materials and Environmental Chemistry Chemical Research Center, Hungarian Academy of Sciences 2006 1 1. INTRODUCTION AND AIMS Widespread applicapibility and the increased quality requirements of permanganate salts require development of new synthetic methods. The transition metal permanganates are precursors of numerous catalytically active metal-manganese oxide composites. Therefore, the exact kowledge of their structure, reactivity and the thermal decomposition characteristics of permanganate compounds is essenial to plan the properties of the decomposition products. The main target of this work was the development of new synthetic methods to prepare simple and complex salts of permanganic acid, including the preparation and the study of ammonium permanganate and some ammine-complex permanganates of transition metals such as Ag, Cu, Ni, Cd, Zn. 2. ANALYTICAL METHODS Classical inorganic analytical chemistry, IR and Raman spectroscopy, UV-VIS spectroscopy, ESR, XRD, GC-MS, single crystal X-ray and powder neutron diffractometry, TG-gas- titrimetry, TG-MS and DSC methods were used. 3. NEW RESULTS 3.1. SYNTHESES 3.1.1. Syntheses based on manganese heptoxide A method has been developed to prepare Ba, Mg, Ca, Ni, Zn, Cd, Cu(II), Al, Fe(III), Y, Ce(IV), Sm, Pr and Gd-permanganates by means of the reaction of KMnO4 with sulfuric acid monohydrate, followed by extraction of the Mn2O7 with CCl4 and the reaction of the Mn2O7 in CCl4 with catalytic amount of water and the oxide, hydroxide and carbonate of the appropriate metal. 3.1.2. Syntheses based on the metathesis reaction of KMnO4 Reaction of warm saturated solutions of NH4Cl and potassium permangnate provided Kx(NH4)1-xMnO4 (x=0.2-0.4) solid solutions instead of NH4MnO4 as it has been declared previously. -

Prohibited and Restricted Chemical List
School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List PROHIBITED AND RESTRICTED CHEMICAL LIST Introduction After incidents of laboratory chemical contamination at several schools, DCPS, The American Association for the Advancement of Science (AAAS) and DC Fire and Emergency Management Services developed an aggressive program for chemical control to eliminate student and staff exposure to potential hazardous chemicals. Based upon this program, all principals are required to conduct a complete yearly inventory of all chemicals located at each school building to identify for the removal and disposal of any prohibited/banned chemicals. Prohibited chemicals are those that pose an inherent, immediate, and potentially life- threatening risk, injury, or impairment due to toxicity or other chemical properties to students, staff, or other occupants of the school. These chemicals are prohibited from use and/or storage at the school, and the school is prohibited from purchasing or accepting donations of such chemicals. Restricted chemicals are chemicals that are restricted by use and/or quantities. If restricted chemicals are present at the school, each storage location must be addressed in the school's written emergency plan. Also, plan maps must clearly denote the storage locations of these chemicals. Restricted chemicals—demonstration use only are a subclass in the Restricted chemicals list that are limited to instructor demonstration. Students may not participate in handling or preparation of restricted chemicals as part of a demonstration. If Restricted chemicals—demonstration use only are present at the school, each storage location must be addressed in the school's written emergency plan. Section 7: Appendices – October 2009 37 School Emergency Response Plan and Management Guide Prohibited and Restricted Chemical List Following is a table of chemicals that are Prohibited—banned, Restricted—academic curriculum use, and Restricted—demonstration use only. -

Basic Description for Ground and Air Hazardous
BASIC DESCRIPTION FOR GROUND AND AIR GROUND AND AIR HAZARDOUS MATERIALS SHIPMENTS GROUND SHIPMENTS AIR SHIPMENTS SHIPMENTS HAZARD DOT DOT CLASS OR MAXIMUM EXEMPTION, GROUND EXEMPTION, HAZARDOUS MATERIALS DESCRIPTIONS DIVISION I.D. NUMBER LABEL(S) REQUIRED OR QUANTITY PER SPECIAL SERVICE TO LABEL(S) REQUIRED OR MAXIMUM NET CARGO SPECIAL NON-BULK AND PROPER SHIPPING NAME (Subsidiary if (ALSO MARK PACKING EXEMPTION, SPECIAL PERMIT INNER PERMIT CANADA EXEMPTION, SPECIAL PERMIT QUANTITY PER AIRCRAFT PERMIT SPECIAL EXCEPTIONS PACKAGING (ALSO MARK ON PACKAGE) applicable) ON PACKAGE) GROUP OR EXCEPTION RECEPTACLE OR 173.13 PERMITTED OR EXCEPTION PACKAGE** QUANTITY OR 173.13 PROVISIONS §173.*** §173.*** (1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) Accellerene, see p-Nitrosodimethylaniline Accumulators, electric, see Batteries, wet etc Accumulators, pressurized, pneumatic or hydraulic (containing non-flammable gas), see Articles pressurized, pneumatic or hydraulic (containing non-flammable gas) FLAMMABLE FLAMMABLE Acetal 3 UN1088 II LIQUID * YES LIQUID * 5 L 150 202 FLAMMABLE Acetaldehyde 3 UN1089 I LIQUID YES Forbidden None 201 May not be regulated when shipped via UPS Acetaldehyde ammonia 9 UN1841 III ground YES CLASS 9 * 30 kg 30 kg 155 204 FLAMMABLE FLAMMABLE Acetaldehyde oxime 3 UN2332 III LIQUID * YES LIQUID * 25 L 150 203 CORROSIVE, CORROSIVE, Acetic acid, glacial or Acetic acid solution, FLAMMABLE FLAMMABLE A3, A6, with more than 80 percent acid, by mass 8 (3) UN2789 II LIQUID * YES LIQUID * 1 L A7, A10 154 202 Acetic -

The True Formula
ON THE TRUE FORMULA FOR PERMANGANATES. DISSERTATION PRESENTED TO THE FACULTY OF THE UNIVERSITT7 ' OF VIRGINIA. In application fir the Degree of Dadar of 'Pflilawplzj'. BY CHARLES M. B‘RADBURY, 0F VIRGINIA. THESIS —Permanga;zz}: acid is moiwéaszc, and the per-:V _ mangamztes are represented 6}! the generalfbmwla R (Mn 04 p. -' : . ; V To His Fat/tor, 31m ‘18. granary, (35511. 77:13 Paper is Inscribed IN TOKEN OF AFFECTION, BY film 9mm, Preface. The greater part of the following paper was written nearly a year ago. As it was nearing completion, I found, from an allusion in .an article by M. Raoult, of Paris, (see Bzzlleiz'n dc Soc. C/u'm. de Paris, XLVI, p. 805), that my conclusion as to the true formula for permanganates had been reached already by that eminent chemist, by means of a new method, origi- nated by himself, for determining molecular weights. Upon examining M. Raoult‘s original papem‘fHéWIz—Iifilc C/zz'm. ct dc Ply/5., (6), VIII, July, 1886, p. 3 30, however, it was found that nothing was given concerning permanganates, beyond the simple statement that they were shown by the method to be monobasic. T Dr. Victor Meyer and Dr. Auwers, of Gottingen, have recently extended and simplified the method of M. Raoult, (see Berk/zit a’n’ Dads. Chem. 656115., for February 27 and March 12, 1888), and I am therefore enabled to add materially to the evidence already collected in the dissertation, by a con- vincing application of this method. ‘ Had the detailed results of M. -

LABORATORY SAFETY and CHEMICAL HYGIENE PLAN
LABORATORY SAFETY and CHEMICAL HYGIENE PLAN INDIANA UNIVERSITY Produced by University Environmental Health and Safety December 1996 in cooperation with University Chemical Hygiene Committees Revised 2015 Page 1 Table of Contents EMERGENCY INFORMATION .................................................................................................. 4 Major Emergencies............................................................................................................ 4 Minor Emergencies............................................................................................................ 4 Accident Reports ............................................................................................................... 4 Fires and Fire Alarms ........................................................................................................ 5 Tornado Watches and Warnings (Sirens) .......................................................................... 6 LABORATORY SAFETY CONTACTS ...................................................................................... 7 1. INTRODUCTION ........................................................................................................10 1.1. Purpose ................................................................................................................ 10 1.2. Regulatory Basis ................................................................................................... 10 1.3. Applicability .......................................................................................................... -

Patent Office
Patented May 2, 1939 2,156,727 UNITED STATES PATENT OFFICE 2,156,72 BLUE PIGMENT COLORS AND A PROCESS OF PREPARNG THEV Erich Korinth, Frankfort-on-the-Main-Hochst, and Georg Medler, Murasier, Germany, assign ors to H. G. Faroenindustrie Aktiengesellschaft, Frankfort-on-the-Main, Germany No Drawing. Application August 31, 1934, Se ria No. 42,346. in Germany September 13, 933 Clains. (C. 134-58) The present invention relates to blue pigment fate to run into an aqueous suspension of barium colors prepared from barium Sulfate and to a carbonate which simultaneously contains the proceSS of preparing them. Salts of manganese in a dissolved condition. The We have found that a new kind of stable blue precipitate then contains a sufficient quantity of s pigment colors can be prepared in an economical manganese compounds, so that in the Subsequent nanner from barium Sulfate With the aid of man heating process the desired blue color may be ganese. COnpounds. These pigment colors fulfil obtained. It is particularly advantageous fur the highest requirements. thermore to add a Substance having a basic ac The characteristic feature of the new proceSS tion, for instance barium carbonate and to carry O consists in heating barium Sulfate together With Out the process in an oxidizing melt; the blue O manganese compounds So as to obtain a thermic pigment thus produced has a particularly deep reaction, under Such conditions as lead to the color. production of the MnO4 group or counteract itS The barium sulfate may also be produced only decomposition. This can be attained, in a man during the heating process, for instance from 5 ner in itself known, by carrying out the heating barium carbonate or barium nitrate and sodium 5 process in the presence of basic Substances and Sulfate. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -
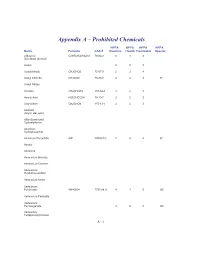
Appendix a – Prohibited Chemicals
Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special 2-Butanol C2H5CH(OH)CH3 78-92-2 0 1 3 (Sec-Butyl Alcohol) Acetal 023 Acetaldehyde CH3CHO5 75-07-0 2 3 4 Acetyl Chloride CH3COCl 75-36-5 2 3 3 W Acetyl Nitrate Acrolein CH2CHCHO 107-02-8 3 4 3 Acrylic Acid H2CCHCO2H 79-10-7 2 2 2 Acrylonitrile CH2CHCN 107-13-1 2 4 3 Alcohols (Allylic, Benzylic) Alkly-Substituted Cycloaliphatics Aluminum Hydrophosphide Aluminum Phosphide AlP 20859-73- 2 4 4 W Amatol Ammonal Ammonium Bromate Ammonium Chlorate Ammonium Hexanitrocobaltate Ammonium Nitrite Ammonium Perchlorate NH4ClO4 7790-98-9 4 1 0 OX Ammonium Periodate Ammonium Permanganate 300 OX Ammonium Tetraperoxychromate A - 1 Appendix A – Prohibited Chemicals NFPA NFPA NFPA NFPA Name Formula CAS #Reactive Health Flammable Special Antimony Compounds Arsenic And Arsenic Compounds Azides Azidocarbonyl Guanidine Barium Ba 2 2 1 W Barium Chlorate Ba(ClO3)2*H2O 13477-00- 1 2 0 OX Barium Oxide (Anhydrous) BaO 1304-28-5 2 3 0 Barium Peroxide BaO2 1304-29-6 0 1 0 OX Benzene C6H6 71-43-2 0 2 3 Benzene Diazonium Chloride Benzotriazole C6H5N3 95-14-7 0 2 1 Benzoyl Peroxide (C6H5CO)2O2 94-36-0 4 1 4 OX Benzyl Alcohol C6H5CH2OH 100-51-6 0 2 1 Bismuth Nitrate Bi(NO3)3*5H2O 10035-06- 3 1 0 OX Borane,Boranes, Diboranes Boron Tribromide 230 W Boron Trifluoride 140 Bromine Pentafluoride Brf5 7789-30-2 3 4 0 W,O Bromine Trifluoride 3 4 0 W,O Butadiene C4H6/CH2=(CH)2=CH 106-99-0 0 2 4 Butenetroil Trinitrate Cadmium and Cadmium Compounds Calcium Nitrate, Anhydrous Ca(NO3)2 -

2020 Emergency Response Guidebook
2020 A guidebook intended for use by first responders A guidebook intended for use by first responders during the initial phase of a transportation incident during the initial phase of a transportation incident involving hazardous materials/dangerous goods involving hazardous materials/dangerous goods EMERGENCY RESPONSE GUIDEBOOK THIS DOCUMENT SHOULD NOT BE USED TO DETERMINE COMPLIANCE WITH THE HAZARDOUS MATERIALS/ DANGEROUS GOODS REGULATIONS OR 2020 TO CREATE WORKER SAFETY DOCUMENTS EMERGENCY RESPONSE FOR SPECIFIC CHEMICALS GUIDEBOOK NOT FOR SALE This document is intended for distribution free of charge to Public Safety Organizations by the US Department of Transportation and Transport Canada. This copy may not be resold by commercial distributors. https://www.phmsa.dot.gov/hazmat https://www.tc.gc.ca/TDG http://www.sct.gob.mx SHIPPING PAPERS (DOCUMENTS) 24-HOUR EMERGENCY RESPONSE TELEPHONE NUMBERS For the purpose of this guidebook, shipping documents and shipping papers are synonymous. CANADA Shipping papers provide vital information regarding the hazardous materials/dangerous goods to 1. CANUTEC initiate protective actions. A consolidated version of the information found on shipping papers may 1-888-CANUTEC (226-8832) or 613-996-6666 * be found as follows: *666 (STAR 666) cellular (in Canada only) • Road – kept in the cab of a motor vehicle • Rail – kept in possession of a crew member UNITED STATES • Aviation – kept in possession of the pilot or aircraft employees • Marine – kept in a holder on the bridge of a vessel 1. CHEMTREC 1-800-424-9300 Information provided: (in the U.S., Canada and the U.S. Virgin Islands) • 4-digit identification number, UN or NA (go to yellow pages) For calls originating elsewhere: 703-527-3887 * • Proper shipping name (go to blue pages) • Hazard class or division number of material 2.