Characterization of New Microsatellite Markers Based on the Transcriptome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi
The University of Southern Mississippi The Aquila Digital Community Honors Theses Honors College Spring 5-2016 Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi Hanna M. Miller University of Southern Mississippi Follow this and additional works at: https://aquila.usm.edu/honors_theses Part of the Biodiversity Commons, and the Botany Commons Recommended Citation Miller, Hanna M., "Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi" (2016). Honors Theses. 389. https://aquila.usm.edu/honors_theses/389 This Honors College Thesis is brought to you for free and open access by the Honors College at The Aquila Digital Community. It has been accepted for inclusion in Honors Theses by an authorized administrator of The Aquila Digital Community. For more information, please contact [email protected]. The University of Southern Mississippi Vascular Flora of the Possum Walk Trail at the Infinity Science Center, Hancock County, Mississippi by Hanna Miller A Thesis Submitted to the Honors College of The University of Southern Mississippi in Partial Fulfillment of the Requirement for the Degree of Bachelor of Science in the Department of Biological Sciences May 2016 ii Approved by _________________________________ Mac H. Alford, Ph.D., Thesis Adviser Professor of Biological Sciences _________________________________ Shiao Y. Wang, Ph.D., Chair Department of Biological Sciences _________________________________ Ellen Weinauer, Ph.D., Dean Honors College iii Abstract The North American Coastal Plain contains some of the highest plant diversity in the temperate world. However, most of the region has remained unstudied, resulting in a lack of knowledge about the unique plant communities present there. -
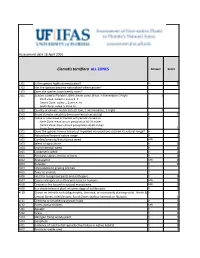
WRA.Datasheet.Template (Version 1) (Version 1).Xlsx
Assessment date 18 April 2016 Clematis terniflora ALL ZONES Answer Score 1.01 Is the species highly domesticated? n 0 1.02 Has the species become naturalised where grown? 1.03 Does the species have weedy races? 2.01 Species suited to Florida's USDA climate zones (0-low; 1-intermediate; 2-high) 2 North Zone: suited to Zones 8, 9 Central Zone: suited to Zones 9, 10 South Zone: suited to Zone 10 2.02 Quality of climate match data (0-low; 1-intermediate; 2-high) 2 2.03 Broad climate suitability (environmental versatility) y 1 2.04 Native or naturalized in habitats with periodic inundation y North Zone: mean annual precipitation 50-70 inches Central Zone: mean annual precipitation 40-60 inches South Zone: mean annual precipitation 40-60 inches 1 2.05 Does the species have a history of repeated introductions outside its natural range? y 3.01 Naturalized beyond native range y 2 3.02 Garden/amenity/disturbance weed unk 3.03 Weed of agriculture n 0 3.04 Environmental weed y 4 3.05 Congeneric weed y 2 4.01 Produces spines, thorns or burrs n 0 4.02 Allelopathic unk 0 4.03 Parasitic n 0 4.04 Unpalatable to grazing animals ? 4.05 Toxic to animals ? 4.06 Host for recognised pests and pathogens n 0 4.07 Causes allergies or is otherwise toxic to humans unk 0 4.08 Creates a fire hazard in natural ecosystems unk 0 4.09 Is a shade tolerant plant at some stage of its life cycle n 0 4.10 Grows on infertile soils (oligotrophic, limerock, or excessively draining soils). -

State of Delaware Invasive Plants Booklet
Planting for a livable Delaware Widespread and Invasive Growth Habit 1. Multiflora rose Rosa multiflora S 2. Oriental bittersweet Celastrus orbiculata V 3. Japanese stilt grass Microstegium vimineum H 4. Japanese knotweed Polygonum cuspidatum H 5. Russian olive Elaeagnus umbellata S 6. Norway maple Acer platanoides T 7. Common reed Phragmites australis H 8. Hydrilla Hydrilla verticillata A 9. Mile-a-minute Polygonum perfoliatum V 10. Clematis Clematis terniflora S 11. Privet Several species S 12. European sweetflag Acorus calamus H 13. Wineberry Rubus phoenicolasius S 14. Bamboo Several species H Restricted and Invasive 15. Japanese barberry Berberis thunbergii S 16. Periwinkle Vinca minor V 17. Garlic mustard Alliaria petiolata H 18. Winged euonymus Euonymus alata S 19. Porcelainberry Ampelopsis brevipedunculata V 20. Bradford pear Pyrus calleryana T 21. Marsh dewflower Murdannia keisak H 22. Lesser celandine Ranunculus ficaria H 23. Purple loosestrife Lythrum salicaria H 24. Reed canarygrass Phalaris arundinacea H 25. Honeysuckle Lonicera species S 26. Tree of heaven Alianthus altissima T 27. Spotted knapweed Centaruea biebersteinii H Restricted and Potentially-Invasive 28. Butterfly bush Buddleia davidii S Growth Habit: S=shrub, V=vine, H=herbaceous, T=tree, A=aquatic THE LIST • Plants on The List are non-native to Delaware, have the potential for widespread dispersal and establishment, can out-compete other species in the same area, and have the potential for rapid growth, high seed or propagule production, and establishment in natural areas. • Plants on Delaware’s Invasive Plant List were chosen by a committee of experts in environmental science and botany, as well as representatives of State agencies and the Nursery and Landscape Industry. -

Mistaken Identity? Invasive Plants and Their Native Look-Alikes: an Identification Guide for the Mid-Atlantic
Mistaken Identity ? Invasive Plants and their Native Look-alikes an Identification Guide for the Mid-Atlantic Matthew Sarver Amanda Treher Lenny Wilson Robert Naczi Faith B. Kuehn www.nrcs.usda.gov http://dda.delaware.gov www.dsu.edu www.dehort.org www.delawareinvasives.net Published by: Delaware Department Agriculture • November 2008 In collaboration with: Claude E. Phillips Herbarium at Delaware State University • Delaware Center for Horticulture Funded by: U.S. Department of Agriculture Natural Resources Conservation Service Cover Photos: Front: Aralia elata leaf (Inset, l-r: Aralia elata habit; Aralia spinosa infloresence, Aralia elata stem) Back: Aralia spinosa habit TABLE OF CONTENTS About this Guide ............................1 Introduction What Exactly is an Invasive Plant? ..................................................................................................................2 What Impacts do Invasives Have? ..................................................................................................................2 The Mid-Atlantic Invasive Flora......................................................................................................................3 Identification of Invasives ..............................................................................................................................4 You Can Make a Difference..............................................................................................................................5 Plant Profiles Trees Norway Maple vs. Sugar -

Determination of Taxonomic Status of Chinese Species of the Genus Clematis by Using High Performance Liquid Chromatography–Mass Spectrometry (Hplc-Ms) Technique
Pak. J. Bot., 42(2): 691-702, 2010. DETERMINATION OF TAXONOMIC STATUS OF CHINESE SPECIES OF THE GENUS CLEMATIS BY USING HIGH PERFORMANCE LIQUID CHROMATOGRAPHY–MASS SPECTROMETRY (HPLC-MS) TECHNIQUE MUHAMMAD ISHTIAQ CH1, 2*, Q. HE2, S. FENG, YI WANG2, P.G. XIAO2, YIYU CHENG2* AND HABIB AHMED3 1Department of Botany, Mirpur University of Science & Technology (MUST) Bhimber Campus, (AK) Pakistan 2Department of Chinese Medicine Science and Engineering, College of Pharmaceutical Sciences, Institute of Bioinformatics, Zhejiang University, No. 38 Zhe da Road Hangzhou, 310027, P. R. China 3Department of Botany, University of Hazara, Garden Campus Mansehra, NWFP, Pakistan. Abstract A comparative taxonomic study using chemometric and numerical taxonomic approaches on 15 populations of 12 species of major taxa of the genus Clematis, belonging to sections, Rectae, Clematis, Meclatis, Tubulosae and Viorna were analyzed by HPLC coupled with diode array detector and ESI-MS. The chemodiversity profile of saponins proved to be useful taxonomic markers for the genus and results are presented in phenograms. The compound ‘Huzhangoside D’ was the most abundant in analyzed species of the genus. Numerical taxonomic study was also conducted based on phylogenetically informative characters which corroborated with chemical fingerprinting findings. The significance of chemical markers in taxonomic study as well as their correlation between morphology and chemical compound profile is also debated with its significant role in botanic drugs identification. Introduction Clematis L., is the second largest genus of Ranunculaceae, which comprises of more than 300 species worldwide and among these 147 (93 endemic) species are found in China (Wang, 1999). Traditional classification systems are usually based on floral and vegetative characters, and have been used for taxonomic divisions of many plants. -

Vascular Plant Inventory and Ecological Community Classification for Cumberland Gap National Historical Park
VASCULAR PLANT INVENTORY AND ECOLOGICAL COMMUNITY CLASSIFICATION FOR CUMBERLAND GAP NATIONAL HISTORICAL PARK Report for the Vertebrate and Vascular Plant Inventories: Appalachian Highlands and Cumberland/Piedmont Networks Prepared by NatureServe for the National Park Service Southeast Regional Office March 2006 NatureServe is a non-profit organization providing the scientific knowledge that forms the basis for effective conservation action. Citation: Rickie D. White, Jr. 2006. Vascular Plant Inventory and Ecological Community Classification for Cumberland Gap National Historical Park. Durham, North Carolina: NatureServe. © 2006 NatureServe NatureServe 6114 Fayetteville Road, Suite 109 Durham, NC 27713 919-484-7857 International Headquarters 1101 Wilson Boulevard, 15th Floor Arlington, Virginia 22209 www.natureserve.org National Park Service Southeast Regional Office Atlanta Federal Center 1924 Building 100 Alabama Street, S.W. Atlanta, GA 30303 The view and conclusions contained in this document are those of the authors and should not be interpreted as representing the opinions or policies of the U.S. Government. Mention of trade names or commercial products does not constitute their endorsement by the U.S. Government. This report consists of the main report along with a series of appendices with information about the plants and plant (ecological) communities found at the site. Electronic files have been provided to the National Park Service in addition to hard copies. Current information on all communities described here can be found on NatureServe Explorer at www.natureserveexplorer.org. Cover photo: Red cedar snag above White Rocks at Cumberland Gap National Historical Park. Photo by Rickie White. ii Acknowledgments I wish to thank all park employees, co-workers, volunteers, and academics who helped with aspects of the preparation, field work, specimen identification, and report writing for this project. -

Clematis Terniflora
Green Gone Bad Featured Ornamental Plant: Clematis terniflora Some exotic ornamental plants behave badly when they escape from the place they are planted. Infestations of these plants have negative impacts on natural environments. One of these plants is Clematis terniflora; common name: sweet autumn virginsbower. Clematis terniflora is a climbing, semi-evergreen, twining ornamental vine which has been observed and recorded growing outside of cultivation in South Carolina, especially near creeks. It can be seen growing wild in urban areas and creeping into adjacent natural communities. Clematis terniflora is in the family Ranunculaceae (buttercup or crowfoot family). It is native to Japan, Korea, and China and has been cultivated as an ornamental plant in the United States since 1877. Alternate common names include leatherleaf clematis, yam-leaved clematis. This species is synonymous with and is sometimes sold as Clematis maximowicziana and Clematis dioscoreifolia. Clematis terniflora leaflet Clematis terniflora leaves/leaflets/stem Clematis terniflora stems climb with tendril-like petioles and leaf rachises. Leaves are opposite, compound, with 3-5 elliptic leathery leaflets that are shiny and able to persist through mild winters. Leaf margins are entire. Leaflets are Clematis each 2-3 inches (5-7.6 cm) long. Mature bark is light brown and terniflora shreds longitudinally. Abundant white, fragrant, seeds four-petaled flowers appear in late summer through the fall. Seeds are also showy and production is prolific. Seed heads have long, silvery-gray, feather-like hairs attached. The problem with Clematis ternifolia is the vigorous, extremely rampant growth habit. If given support, it will climb rapidly with the aid of tendrilous leaf petioles to 20-25 feet in length. -

Clematis Terniflora
NEW YORK NON -NATIVE PLANT INVASIVENESS RANKING FORM Scientific name: Clematis terniflora USDA Plants Code: CLTE4 Common names: Japanese virgin's-bower Native distribution: Eastern Asia Date assessed: February 20, 2009 Assessors: Steve Glenn, Gerry Moore Reviewers: LIISMA SRC Date Approved: 11 Mar. 2009 Form version date: 22 October 2008 New York Invasiveness Rank: High (Relative Maximum Score 70.00-80.00) Distribution and Invasiveness Rank (Obtain from PRISM invasiveness ranking form ) PRISM Status of this species in each PRISM: Current Distribution Invasiveness Rank 1 Adirondack Park Invasive Program Not Assessed Not Assessed 2 Capital/Mohawk Not Assessed Not Assessed 3 Catskill Regional Invasive Species Partnership Not Assessed Not Assessed 4 Finger Lakes Not Assessed Not Assessed 5 Long Island Invasive Species Management Area Widespread High 6 Lower Hudson Not Assessed Not Assessed 7 Saint Lawrence/Eastern Lake Ontario Not Assessed Not Assessed 8 Western New York Not Assessed Not Assessed Invasiveness Ranking Summary Total (Total Answered*) Total (see details under appropriate sub-section) Possible 1 Ecological impact 40 ( 20 ) 10 2 Biological characteristic and dispersal ability 25 ( 22 ) 19 3 Ecological amplitude and distribution 25 ( 25) 22 4 Difficulty of control 10 ( 6) 2 Outcome score 100 ( 73 )b 53a † Relative maximum score 72.60 § New York Invasiveness Rank High (Relative Maximum Score 70.00-80.00) * For questions answered “unknown” do not include point value in “Total Answered Points Possible.” If “Total Answered Points Possible” is less than 70.00 points, then the overall invasive rank should be listed as “Unknown.” †Calculated as 100(a/b) to two decimal places. -

Plant Invaders of Mid-Atlantic Natural Areas Revised & Updated – with More Species and Expanded Control Guidance
Plant Invaders of Mid-Atlantic Natural Areas Revised & Updated – with More Species and Expanded Control Guidance National Park Service U.S. Fish and Wildlife Service 1 I N C H E S 2 Plant Invaders of Mid-Atlantic Natural Areas, 4th ed. Authors Jil Swearingen National Park Service National Capital Region Center for Urban Ecology 4598 MacArthur Blvd., N.W. Washington, DC 20007 Britt Slattery, Kathryn Reshetiloff and Susan Zwicker U.S. Fish and Wildlife Service Chesapeake Bay Field Office 177 Admiral Cochrane Dr. Annapolis, MD 21401 Citation Swearingen, J., B. Slattery, K. Reshetiloff, and S. Zwicker. 2010. Plant Invaders of Mid-Atlantic Natural Areas, 4th ed. National Park Service and U.S. Fish and Wildlife Service. Washington, DC. 168pp. 1st edition, 2002 2nd edition, 2004 3rd edition, 2006 4th edition, 2010 1 Acknowledgements Graphic Design and Layout Olivia Kwong, Plant Conservation Alliance & Center for Plant Conservation, Washington, DC Laurie Hewitt, U.S. Fish & Wildlife Service, Chesapeake Bay Field Office, Annapolis, MD Acknowledgements Funding provided by the National Fish and Wildlife Foundation with matching contributions by: Chesapeake Bay Foundation Chesapeake Bay Trust City of Bowie, Maryland Maryland Department of Natural Resources Mid-Atlantic Invasive Plant Council National Capital Area Garden Clubs Plant Conservation Alliance The Nature Conservancy, Maryland–DC Chapter Worcester County, Maryland, Department of Comprehensive Planning Additional Fact Sheet Contributors Laurie Anne Albrecht (jetbead) Peter Bergstrom (European -

Development of Conservation Targets for the Lower Hudson PRISM (LHP) Final Report: July 1 to September 15, 2014
Development of Conservation Targets for the Lower Hudson PRISM (LHP) Final Report: July 1 to September 15, 2014 John Mickelson Geospatial & Ecological Services 501 Stage Road Monroe NY, 10950-3217 (845) 893-4110, [email protected] Figure 1. Lower Hudson PRISM region Figure 2. Lower Hudson PRISM and adjacent counties Review of Completed Scope of Work: LHP Action Plan Items Addressed: all contracted project tasks have been completed for the LHP Goals and Objectives of the Lower Hudson 2014 Action Plan 2.1 and 2.2 and 3.1, as outlined within the Project Deliverables section (see Appendix A). While active participation and input of the Conservation Target Working Group has remained muted, the consultant has accomplished the following and will continue through with meetings with the LHP Partners Meeting (Oct 1, 2014) and the Conservation Targets working group meeting (November 5, 2014), to continue momentum into the 2015 project year. Specific project accomplishments include. : GOAL 2: CONSERVATION TARGETS STATUS Priority Strategies (ranked): 1. Coordinating Conservation Target Partners 2. Managing IS strategically 3. Assessment and Monitoring Network Objective 2.1 Engage Partners COMPLETED Since the projects inception, research has engaged important and valuable conservation efforts conducted by over 22 regional agencies and programs (see Table 1) that the LHP might benefit from considering when developing Conservation Targets. All of these have been engaged and valuable geospatial & thematic data have been acquired from many of these potential partners. Four agencies feature especially prominently within our region and form the core of the proposed Conservation Target overlay matrices (see Conservation Target section below): • New York State Natural Heritage Program • Hudson River Estuary Program • The Nature Conservancy • New York Audubon Society These groups (collectively referred to hereafter as CTLA) should be prioritized for partnership relationships over the coming years. -

Considérations Sur L'histoire Naturelle Des Ranunculales
Considérations sur l’histoire naturelle des Ranunculales Laetitia Carrive To cite this version: Laetitia Carrive. Considérations sur l’histoire naturelle des Ranunculales. Botanique. Université Paris Saclay (COmUE), 2019. Français. NNT : 2019SACLS177. tel-02276988 HAL Id: tel-02276988 https://tel.archives-ouvertes.fr/tel-02276988 Submitted on 3 Sep 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Considérations sur l’histoire naturelle des Ranunculales 2019SACLS177 Thèse de doctorat de l'Université Paris-Saclay : préparée à l’Université Paris-Sud NNT École doctorale n°567 : Sciences du végétal, du gène à l'écosystème (SDV) Spécialité de doctorat : Biologie Thèse présentée et soutenue à Orsay, le 05 juillet 2019, par Laetitia Carrive Composition du Jury : Catherine Damerval Directrice de recherche, CNRS (– UMR 320 GQE) Présidente du jury Julien Bachelier Professeur, Freie Universität Berlin (– Institute of Biology) Rapporteur Thomas Haevermans Maître de conférences, MNHN (– UMR 7205 ISYEB) Rapporteur Jean-Yves Dubuisson Professeur, SU (–UMR 7205 ISYEB) Examinateur Sophie Nadot Professeure, U-PSud (– UMR 8079 ESE) Directrice de thèse « Le commencement sera d’admirer tout, même les choses les plus communes. Le milieu, d’écrire ce que l’on a bien vu et ce qui est d’utilité. -
Two New Eriophyid Mite Species Associated with Clematis Terniflora Var
A peer-reviewed open-access journal ZooKeys 621:Two 1–14 new (2016) eriophyid mite species associated with Clematis terniflora var. mandshurica... 1 doi: 10.3897/zookeys.621.9443 RESEARCH ARTICLE http://zookeys.pensoft.net Launched to accelerate biodiversity research Two new eriophyid mite species associated with Clematis terniflora var. mandshurica in China (Acari, Eriophyidae) Yan Dong1, Yan-Mei Sun2, Xiao-Feng Xue1 1 Department of Entomology, Nanjing Agricultural University, Nanjing, Jiangsu 210095, China 2 Department of Plant Protection, Jilin Agricultural Science and Technology University, Jilin, Jilin 132101, China Corresponding author: Xiao-Feng Xue ([email protected]) Academic editor: E. de Lillo | Received 5 June 2016 | Accepted 20 September 2016 | Published 3 October 2016 http://zoobank.org/165599AE-3457-486D-8C0D-6B2EB54291E0 Citation: Dong Y, Sun Y-M, Xue X-F (2016) Two new eriophyid mite species associated with Clematis terniflora var. mandshurica in China (Acari, Eriophyidae). ZooKeys 621: 1–14. doi: 10.3897/zookeys.621.9443 Abstract Two new eriophyid mite species associated with Clematis ternifloravar. mandshurica, namely Aculops jilinensis sp. n. and Phyllocoptes terniflores sp. n., are described. Both species infest the tender leaves of host plants, inducing severe curling and blistering. Keywords Aculops, Phyllocoptes, plant feeding, taxonomy Introduction Clematis ternifloraDC. var. mandshurica (Rupr.) Ohwi, called “la liao tie xian lian” in Mandarin, belongs to the family Ranunculaceae, and is native to China (Heilongji- ang, Jilin, Liaoning, Shanxi, Inner Mongolia), Korea, Mongolia, and the Russian Far East (website of Flora Republicae Popularis Sinicae – http://frps.eflora.cn/frps/Clema- tis%20terniflora and Germplasm Resources Information Network – https://npgsweb.